Síndrome de Prader-Willi
Síndrome de Prader-Willi | |
|---|---|
 Criança de oito anos com síndrome de Prader-Willi,[1] mostrando obesidade característica | |
Especialidade | genética médica, pediatria, neurologia |
| Classificação e recursos externos | |
CID-10 | Q87.1 |
CID-9 | 759.81 |
OMIM | 176270 |
DiseasesDB | 10481 |
MedlinePlus | 001605 |
eMedicine | ped/1880 |
MeSH | D011218 |
A síndrome de Prader-Willi (PWS) é uma desordem genética devido à perda de função de genes específicos.[2] Em recém-nascidos, os sintomas incluem músculos fracos, má alimentação e desenvolvimento lento. Na infância, a criança fica constantemente com fome, o que muitas vezes leva à obesidade e diabetes tipo 2. Também há tipicamente deficiência intelectual leve a moderada e problemas comportamentais. Muitas vezes, a testa é estreita, as mãos e os pés pequenos, a altura baixa, a cor da pele clara. O portador também é incapaz de ter filhos.[2]
Cerca de 70% dos casos ocorrem quando parte do cromossomo 15 do pai é apagada. Em 25% dos casos, a pessoa tem duas cópias do cromossomo 15 da mãe e nenhuma do pai. Como partes do cromossomo da mãe são desligadas, eles terminam sem cópias operantes de certos genes. A síndrome de Prader-Willi geralmente não é herdada. Em vez disso, as alterações genéticas acontecem durante a formação do ovo ou do esperma ou no desenvolvimento precoce.[2] Não há fatores de risco conhecidos. Aqueles que têm um filho com síndrome de Prader-Willi possuem menos de 1% de chance de ter um outro filho com a síndrome.[3] Um mecanismo semelhante ocorre na síndrome de Angelman, com a diferença de que há um cromossomo 15 defeituoso da mãe ou duas cópias do pai.[4][5]
A síndrome de Prader-Willi não tem cura. O tratamento, entretanto, pode melhorar os sintomas, especialmente se realizado precocemente.[6] Nos recém-nascidos, as dificuldades de alimentação podem ser aliviadas com tubos de alimentação. Uma supervisão estrita da nutrição é normalmente exigida a partir de cerca de três anos em combinação com um programa de exercícios. A terapia com hormônio de crescimento também melhora os sintomas. A terapia comportamental e a medicação podem ajudar com alguns problemas comportamentais. Comunidades terapêuticas são muitas vezes necessárias na idade adulta.[7]
A síndrome de Prader-Willi afeta uma entre 10 mil e 30 mil pessoas.[2] Homens e mulheres são afetados igualmente.[3] A condição recebe este nome em homenagem a Andrea Prader, Heinrich Willi e Alexis Labhart que a descreveram em detalhes em 1956.[8] Uma descrição anterior ocorreu em 1887 por John Langdon.[9][10]
 Reproduzir conteúdo
Reproduzir conteúdoExplicação de vídeo
Índice
1 Sinais e sintomas
1.1 Útero e nascimento
1.2 Infância
1.3 Idade adulta
1.4 Aparência física
1.5 Aspectos neuro-cognitivos
1.6 Aspectos comportamentais
1.7 Aspectos endócrinos
1.8 Aspectos oftalmológicos
2 Genética
3 Diagnóstico
4 Tratamento
5 Epidemiologia
6 Sociedade e Cultura
7 Veja também
8 Referências
Sinais e sintomas |
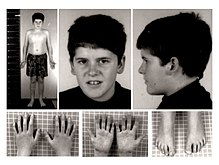
Fenótipo da Síndrome de Prader-Willi em um jovem de 15 anos. Observa-se a ausência de características faciais típicas da Síndrome de Prader–Willi e a presença de leve obesidade truncal.
Existem muitos sinais e sintomas da síndrome de Prader-Willi. Os sintomas podem variar de tônus muscular baixo durante a infância a problemas comportamentais na primeira infância. Outro sintoma geralmente encontrado em bebês, além do tônus reduzido, é a falta de coordenação do olho. Alguns nascem com olhos em forma de amêndoas. Devido ao tônus muscular baixo, o bebê pode não ter um forte reflexo de sucção. Seu choro é baixo e eles têm dificuldade em acordar. Outro sinal desta condição é o lábio superior fino.
Outros aspectos clinicamente observados incluem hipotonia, função neurológica anormal, hipogonadismo, atrasos em cognição e desenvolvimento, hiperfagia, obesidade, baixa estatura e distúrbios comportamentais e psiquiátricos.[11]
V. A. Holm et al. descrevem as seguintes características e sinais como indicadores pré-teste da síndrome de Prader–Willi, embora nem todos estejam presentes.[12]
Útero e nascimento |
- Redução do movimento fetal;
- Posição fetal anormal frequente;
Poli-hidrâmnio ocasional (excesso de líquido amniótico);- Partos sem apresentação cefálica (bebê virado) e cesarianas;
Letargia;
Hipotonia;- Dificuldades de alimentação (o tônus muscular baixo afeta o reflexo de sucção);
- Dificuldades de respiração;
Hipogonadismo.
Infância |
- Transições atrasadas, atraso intelectual;
Sono excessivo;
Estrabismo;
Escoliose (muitas vezes não detectada no nascimento);
Criptorquia (testículos ocultos);- Atraso na fala;
- Má coordenação física;
Hiperfagia (comer demais), que começa entre dois e oito anos e continua em toda a idade adulta. A mudança é notada a partir das dificuldades alimentares na infância;
Ganho excessivo de peso;- Distúrbios do sono;
Puberdade atrasada;- Baixa estatura;
Obesidade;
Flexibilidade extrema.
Idade adulta |
Infertilidade (homens e mulheres)- Hipogonadismo;
- Pelos púbicos esparsos;
- Obesidade;
Hipotonia;- Dificuldades de aprendizagem e funcionamento intelectual limítrofe (mas alguns casos de inteligência média);
- Propensão a diabetes mellitus;
- Flexibilidade extrema.
Aparência física |
- Proeminente ponte nasal;
- Mãos e pés pequenos com dedos cônicos;
- Pele macia, facilmente lesionada;
- Excesso de gordura, especialmente na porção central do corpo;
- Testa alta e estreita;
- Lábio superior fino;
- Boca para baixo;
- Olhos amendoados;
- Pele e cabelo claros em relação a outros familiares;
- Falta de desenvolvimento sexual completo;
Dermatotilexomania (vontade de causar ou agravar lesões da própria pele);
Estrias;- Desenvolvimento motor retardado.
Aspectos neuro-cognitivos |
Indivíduos com síndrome de Prader–Willi estão sujeitos a dificuldades de aprendizagem e atenção. L.M. Curfs e J.P. Fryns conduziram pesquisas sobre diferentes graus de dificuldade de aprendizagem encontrados em pessoas com síndrome de Prader–Willi.[13] Seus resultados, usando uma medida de QI, foram os seguintes:
- 5%: QI acima de 85 (inteligência média baixa a alta);
- 27%: QI entre 70 e 85 (funcionamento intelectual limítrofe);
- 39%: QI entre 50 e 70 (deficiência intelectual leve);
- 27%: QI entre 35 e 50 (deficiência intelectual moderada);
- 1%: QI entre 20 e 35 (deficiência intelectual grave);
- <1%: QI abaixo de 20 (deficiência intelectual profunda).
S.B. Cassidy descobriu que 40% dos indivíduos com síndrome de Prader–Willi têm inteligência limítrofe/média baixa,[14] um número superior aos 32% encontrados no estudo de Curfs e Fryns.[13] No entanto, ambos os estudos sugerem que a maioria dos indivíduos (50-65%) está na faixa entre deficiência intelectual leve e inteligência média baixa.
Crianças com síndrome de Prader–Willi mostram um perfil cognitivo incomum. Muitas vezes, são fortes em organização e percepção visual, incluindo leitura e vocabulário, mas sua capacidade verbal (por vezes afetada pela hipernasalidade) é geralmente mais pobre do que a sua compreensão. Uma habilidade marcada em completar quebra-cabeças tem sido notada,.[13] mas isso pode ser um efeito de prática aumentada.[15]
O processamento de informação auditiva e o processamento sequencial são relativamente pobres, assim como as habilidades em aritmética e escrita, as memórias de trabalho visual e auditiva e o limiar de atenção auditivo. Estes atributos às vezes melhoram com a idade, mas déficits nessas áreas permanecem ao longo da vida adulta.[16]
Aspectos comportamentais |
A síndrome de Prader-Willi é freqüentemente associada a um apetite insaciável, voraz, extremo e constante, que persiste não importa o quanto o paciente come, muitas vezes resultando em obesidade mórbida. Responsáveis pelos pacientes precisam limitar estritamente seu acesso a alimentos, geralmente instalando fechaduras em refrigeradores e nos armários e gabinetes em que os alimentos são armazenados.[17] É a causa genética mais comum de obesidade mórbida em crianças.[18] Atualmente, não há consenso quanto à causa deste sintoma, embora anormalidades genéticas no cromossomo 15 interrompam o funcionamento normal do hipotálamo.[19] Dado que o núcleo arqueado do hipotálamo regula muitos processos básicos, incluindo o apetite, pode haver uma ligação. No hipotálamo de pessoas com síndrome de Prader–Willi, as células nervosas que produzem ocitocina, considerada um hormônio que contribui para a saciedade, são anormais.
Pessoas com síndrome de Prader-Willi têm altos níveis de grelina, o que provavelmente contribui diretamente para o aumento do apetite, a hiperfagia e a obesidade observadas nesta síndrome.[20] Cassidy fala da necessidade de uma descrição clara das expectativas comportamentais, o reforço dos limites comportamentais e o estabelecimento de rotinas regulares.
As principais dificuldades de saúde mental experimentadas por pessoas com síndrome de Prader–Willi incluem comportamento compulsivo (geralmente manifestado na dermatotilexomania) e ansiedade.[16][21] Sintomas psiquiátricos, como alucinações, paranóia e depressão, foram descritos em alguns casos e afetam entre 5 e 10% dos adultos jovens.[16] Os pacientes também são frequente e extremamente teimosos e propensos à raiva.[17] Problemas psiquiátricos e comportamentais são a causa mais comum de hospitalização.[22]
Entre 70 e 90% dos indivíduos afetados desenvolvem padrões comportamentais na primeira infância. Aspectos destes padrões podem incluir teimosia, birras, comportamento controlador e manipulador, dificuldades com mudança de rotina e hábitos compulsivos.[23]
Aspectos endócrinos |
Vários aspectos corroboram a hipótese de que indivíduos com síndrome de Prader–Willi têm deficiência de hormônio do crescimento. Especificamente, pessoas com síndrome de Prader–Willi têm baixa estatura, são obesos com composição corporal anormal, possuem reduzidas massa livre de gordura, massa corporal magra, gasto energético total e densidade óssea.
A síndrome de Prader–Willi é caracterizada pelo hipogonadismo. Isto se manifesta nos testículos que não descem nos homens e na adrenarca prematura benigna nas mulheres. Os testículos podem descer com o tempo ou ser deslocados com cirurgia ou reposição de testosterona. A adrenarca pode ser tratada com terapia de reposição hormonal.
Aspectos oftalmológicos |
A síndrome de Prader–Willi é comumente associada com o desenvolvimento de estrabismo. Em um estudo, mais de 50% dos pacientes apresentaram estrabismo, principalmente esotropia.
Genética |

A síndrome de Prader–Willi é uma desordem causada por um fenômeno epigenético conhecido como imprinting. É provocada pela deleção das cópias paternas dos genes SNRPN e necdin juntamente com clusters de snoRNAs: SNORD64, SNORD107, SNORD108 e duas cópias de SNORD109, 29 cópias de SNORD116 (HBII-85) e 48 cópias de SNORD115 (HBII-52). Estes estão no cromossomo 15 localizado na região 15q11-13.[24][25][26][27] Esta chamada região PWS/AS pode ser perdida por um dos vários mecanismos genéticos que, na maioria dos casos, ocorre através de mutação fortuita. Outros mecanismos menos comuns incluem: dissomia uniparental, mutações esporádicas, translocações cromossômicas, deleções de genes. Devido a imprinting, as cópias herdadas maternalmente destes genes são virtualmente silenciosas, somente as cópias paternas dos genes são expressadas.[28][29] A PWS resulta da perda de cópias paternas desta região. A deleção da mesma região no cromossomo materno causa a síndrome de Angelman (AS). PWS e AS representam os primeiros casos relatados de transtornos de impressão em seres humanos.
O risco para o irmão de uma criança afetada de ter PWS depende do mecanismo genético que causou a doença. O risco para os irmãos é <1% se a criança afetada tiver deleção gênica ou dissomia uniparental, até 50% se a criança afetada tem uma mutação da região de controle de impressão e até 25% se houver translocação cromossômica parental. O teste pré-natal é possível para qualquer dos mecanismos genéticos conhecidos.
Uma microdeleção em uma família do snoRNA HBII-52 tem excluído de desempenhar um papel importante na doença.[30]
Estudos de sistemas de modelos humanos e de ratos mostraram a deleção das 29 cópias da caixa C/D snoRNA SNORD116 (HBII-85) para ser a principal causa da síndrome de Prader-Willi.[31][32][33][34][35]
Diagnóstico |
É tradicionalmente caracterizada por hipotonia, baixa estatura, hiperfagia, obesidade, problemas comportamentais (especificamente comportamentos semelhantes a TOC), mãos e pés pequenos, hipogonadismo e leve incapacidade intelectual.[36] No entanto, com o diagnóstico precoce e tratamento precoce (como com terapia de hormônio do crescimento), o prognóstico para pessoas com PWS está começando a mudar. Como o autismo, PWS é um distúrbio do espectro e os sintomas podem variar de leve a grave e pode mudar ao longo da vida da pessoa. Vários sistemas de órgãos são afetados.
Tradicionalmente, a síndrome de Prader-Willi foi diagnosticada pela apresentação clínica. Atualmente, a síndrome é diagnosticada através de testes genéticos; O teste é recomendado para recém-nascidos com hipotonia pronunciada. O diagnóstico precoce de PWS permite a intervenção precoce, bem como a prescrição precoce do hormônio do crescimento. As injecções diárias de hormona de crescimento recombinante (GH) estão indicadas para crianças com PWS. GH apoia o crescimento linear e aumento da massa muscular, e pode diminuir a preocupação alimentar e ganho de peso.
O pilar do diagnóstico é o teste genético, especificamente o teste de metilação baseado no DNA para detectar a ausência da síndrome de Prader-Willi/Síndrome de Angelman (PWS / AS), contribuída paternalmente, no cromossomo 15q11-q13. Tais testes detectam mais de 97% dos casos. Testes específicos de metilação são importantes para confirmar o diagnóstico de PWS em todos os indivíduos, mas especialmente aqueles que são muito jovens para demonstrar características suficientes para fazer o diagnóstico por motivos clínicos ou nos indivíduos com achados atípicos.
A síndrome de Prader-Willi é muitas vezes diagnosticada erroneamente como outras síndromes devido a muitos na comunidade médica não familiaridade com PWS.[18] Às vezes, é diagnosticado como Síndrome de Down, simplesmente por causa da freqüência relativa da síndrome de Down em comparação com PWS.[18]
Tratamento |
A síndrome de Prader-Willi não tem cura; no entanto, vários tratamentos estão em vigor para diminuir os sintomas da condição. Durante a infância, os indivíduos devem ser submetidos a terapias para melhorar a força muscular. A fala e a terapia ocupacional também são indicadas. Durante os anos escolares, as crianças beneficiam de um ambiente de aprendizagem altamente estruturado, bem como ajuda extra.[17]
Porque hipotonia pode ser um sintoma de PWS, é vital para fornecer nutrição adequada durante a infância. Também é muito importante para estressar a atividade física em indivíduos com PWS para todas as idades, a fim de otimizar a força e promover um estilo de vida saudável.[37]
A prescrição de injecções diárias de hormona de crescimento recombinante está indicada para crianças com PWS. GH apoia o crescimento linear e aumento da massa muscular, e pode diminuir a preocupação alimentar e ganho de peso.[38][39]
Por causa da obesidade severa, a apneia obstrutiva do sono é uma seqüela comum, e uma máquina positiva da pressão da via aérea é frequentemente necessário. Pode vir um momento em que uma pessoa que tenha sido diagnosticada com PWS pode ter que passar por procedimentos cirúrgicos. Uma cirurgia que provou ser malsucedida para o tratamento da obesidade é bypass gástrico. Os pacientes com síndrome de Prader-Willi têm uma tolerância muito alta à dor; Portanto, eles podem estar experimentando sintomas abdominais significativos, como gastrite aguda, apendicite ou colecistite e não estar ciente dela até mais tarde.
Zafgen, Inc. está atualmente a realizar ensaios de fase 3 de beloranib relacionados com o controlo do peso e apetite dos doentes com síndrome de Prader-Willi,[40][41][42] com 2a resultados mostrando 8,1% de gordura corporal reduzida após 4 semanas (na dose mais elevada do estudo de 1,8 mg) e diminuiu apetite, apesar de um aumento de 50% no subsídio de calorias diárias.[43][44] Em dezembro de 2015, Zafgen interrompeu um ensaio clínico de Fase III para a síndrome de Prader-Willi após a segunda morte do paciente, a fim de determinar se as mortes eram relacionadas ao tratamento.[45]
Comportamento e problemas psiquiátricos devem ser detectados precocemente para obter os melhores resultados. Essas questões são melhores quando tratadas com educação e treinamento dos pais. Às vezes, a medicação é introduzida também. Os agonistas da serotonina têm sido mais eficazes na diminuição dos acessos de raiva e na melhoria da compulsividade.[46]
Epidemiologia |
PWS afeta aproximadamente 1 em 10.000 a 1 em 25.000 recém-nascidos.[36] Há mais de 400.000 pessoas que vivem com PWS em todo o mundo.[47]
Sociedade e Cultura |

(1680) Pintura por Juan Carreno de Miranda de Eugenia Martínez Vallejo. Uma menina presumida a ter a síndrome de Prader-Willi[48]
Apesar da sua raridade, a síndrome de Prader-Willi tem sido frequentemente referenciada na cultura popular, em parte devido ao fascínio que rodeia o apetite insaciável e a obesidade que são sintomas.
A síndrome de Prader-Willi tem sido descrita e documentada várias vezes na televisão. Um indivíduo imaginário com a síndrome de Prader-Willi caracterizada no episódio "cão come o cão" da série de televisão CSI: Investigação da cena do crime, que arejou sobre Novembro 24, 2005.[49] Na mídia BRITÂNICA em julho 2007, o canal 4 arejou um documentário em 2006 chamado Não pode parar de comer, envolvendo a vida cotidiana de duas pessoas com síndrome de Prader-Willi, Joe e Tamara.[50] No episódio de 9 de maio de 2010 de Extreme Makeover: Home Edition, Sheryl Crow ajudou Ty Pennington a reconstruir uma casa para uma família cujo filho mais novo, Ethan Starkweather, estava sofrendo de síndrome de Prader-Willi.[51] No episódio de 22 de março de 2012 de Mystery Diagnosis no canal Discovery Health, Conor Heybach, que tem a síndrome de Prader-Willi, compartilhou sua história de como ele foi diagnosticado com ele.
Em dezembro de 2011, o Taipei Times, em Taiwan, destacou a tragédia de um motorista de táxi que havia se matado e sua filha de nove anos que tinha a condição, no que a polícia descreveu como um "provável assassinato-suicídio".[52]
Veja também |
- Epigenética
Referências
↑ Cortés M, Fanny; Alliende R, M. Angélica; Barrios R, Andrés; Curotto L, Bianca; Santa María V, Lorena; Barraza O, Ximena; Troncoso A, Ledia; Mellado S, Cecilia; Pardo V, Rosa (janeiro de 2005). «[Clinical, genetic and molecular features in 45 patients with Prader-Willi syndrome]». Revista Medica De Chile. 133 (1): 33–41. ISSN 0034-9887. PMID 15768148. Consultado em 26 de junho de 2017
↑ abcd Reference, Genetics Home. «Prader-Willi syndrome». Genetics Home Reference (em inglês). Consultado em 26 de junho de 2017
↑ ab «How many people are affected/at risk for Prader-Willi syndrome (PWS)?». www.nichd.nih.gov (em inglês). Consultado em 26 de junho de 2017
↑ «Prader-Willi Syndrome (PWS): Other FAQs». www.nichd.nih.gov (em inglês). Consultado em 26 de junho de 2017
↑ Reference, Genetics Home. «Angelman syndrome». Genetics Home Reference (em inglês). Consultado em 26 de junho de 2017
↑ «Is there a cure for Prader-Willi syndrome (PWS)?». www.nichd.nih.gov (em inglês). Consultado em 26 de junho de 2017
↑ «What are the treatments for Prader-Willi syndrome (PWS)?». www.nichd.nih.gov (em inglês). Consultado em 26 de junho de 2017
↑ «Whonamedit - dictionary of medical eponyms». www.whonamedit.com (em inglês). Consultado em 26 de junho de 2017
↑ Mia, Md Mohan (6 de abril de 2016). Classical and Molecular Genetics (em inglês). [S.l.]: American Academic Press. ISBN 9781631817762
↑ Jorde, Lynn B.; Carey, John C.; Bamshad, Michael J. (12 de agosto de 2015). Medical Genetics E-Book (em inglês). [S.l.]: Elsevier Health Sciences. ISBN 9780323188371
↑ Cassidy, Suzanne B.; Driscoll, Daniel J. (10 de setembro de 2008). «Prader–Willi syndrome». European Journal of Human Genetics (em inglês). 17 (1): 3–13. ISSN 1018-4813. PMID 18781185. doi:10.1038/ejhg.2008.165
↑ Holm, V. A.; Cassidy, S. B.; Butler, M. G.; Hanchett, J. M.; Greenswag, L. R.; Whitman, B. Y.; Greenberg, F. (02 d.C.). «Prader-Willi syndrome: consensus diagnostic criteria». Pediatrics. 91 (2): 398–402. ISSN 0031-4005. PMID 8424017
↑ abc Curfs, L. M.; Fryns, J. P. (1992). «Prader-Willi syndrome: a review with special attention to the cognitive and behavioral profile». Birth Defects Original Article Series. 28 (1): 99–104. ISSN 0547-6844. PMID 1340242
↑ Cassidy, S. B. (1 de novembro de 1997). «Prader-Willi syndrome.». Journal of Medical Genetics (em inglês). 34 (11): 917–923. ISSN 0022-2593. PMID 9391886. doi:10.1136/jmg.34.11.917
↑ Whittington, J.; Holland, A.; Webb, T.; Butler, J.; Clarke, D.; Boer, H. (1 de fevereiro de 2004). «Cognitive abilities and genotype in a population-based sample of people with Prader-Willi syndrome». Journal of intellectual disability research: JIDR. 48 (Pt 2): 172–187. ISSN 0964-2633. PMID 14723659
↑ abc Udwin O (Novembro de 1998). «Prader-Willi syndrome: Psychological and behavioural characteristics». Contact a Family
↑ abc «What are the treatments for Prader-Willi syndrome (PWS)?». www.nichd.nih.gov (em inglês). Consultado em 16 de maio de 2017
↑ abc Nordqvist (Dezembro de 2012). «What Is Prader-Willi Syndrome? What Causes Prader-Willi Syndrome?». Medical News Today. MediLexicon International
↑ Cassidy, S. B. (1 de novembro de 1997). «Prader-Willi syndrome». Journal of Medical Genetics. 34 (11): 917–923. ISSN 0022-2593. PMID 9391886
↑ Cummings, David E.; Clement, Karine; Purnell, Jonathan Q.; Vaisse, Christian; Foster, Karen E.; Frayo, R. Scott; Schwartz, Michael W.; Basdevant, Arnaud; Weigle, David S. «Elevated plasma ghrelin levels in Prader–Willi syndrome». Nature Medicine. 8 (7): 643–644. doi:10.1038/nm0702-643
↑ Clark DJ, Boer H, Webb T (1995). «General and behavioural aspects of PWS: a review». Mental Health Research. 8 (195): 38–49
↑ Cassidy SB, Devi A, Mukaida C (1994). «Aging in PWS: 232 patients over age 30 years». Proc. Greenwood Genetic Centre. 13: 102–3
↑ Cassidy, Suzanne B.; Driscoll, Daniel J. (10 de setembro de 2008). «Prader–Willi syndrome». European Journal of Human Genetics (em inglês). 17 (1): 3–13. ISSN 1018-4813. PMID 18781185. doi:10.1038/ejhg.2008.165
↑ OMIM 17627
↑ de los Santos, T.; Schweizer, J.; Rees, C. A.; Francke, U. (1 de novembro de 2000). «Small evolutionarily conserved RNA, resembling C/D box small nucleolar RNA, is transcribed from PWCR1, a novel imprinted gene in the Prader-Willi deletion region, which Is highly expressed in brain». American Journal of Human Genetics. 67 (5): 1067–1082. ISSN 0002-9297. PMID 11007541. doi:10.1086/303106
↑ Cavaillé, J.; Buiting, K.; Kiefmann, M.; Lalande, M.; Brannan, C. I.; Horsthemke, B.; Bachellerie, J. P.; Brosius, J.; Hüttenhofer, A. (19 de dezembro de 2000). «Identification of brain-specific and imprinted small nucleolar RNA genes exhibiting an unusual genomic organization». Proceedings of the National Academy of Sciences of the United States of America. 97 (26): 14311–14316. ISSN 0027-8424. PMID 11106375. doi:10.1073/pnas.250426397
↑ "Prader-Willi Syndrome - MeSH - NCBI." National Center for Biotechnology Information. U.S. National Library of Medicine, n.d. Web. 01 Nov. 2016. <https://www.ncbi.nlm.nih.gov/mesh/68011218>.
↑ Buiting, K.; Saitoh, S.; Gross, S.; Dittrich, B.; Schwartz, S.; Nicholls, R. D.; Horsthemke, B. (1 de abril de 1995). «Inherited microdeletions in the Angelman and Prader-Willi syndromes define an imprinting centre on human chromosome 15». Nature Genetics. 9 (4): 395–400. ISSN 1061-4036. PMID 7795645. doi:10.1038/ng0495-395
↑ «Major breakthrough in understanding Prader-Willi syndrome, a parental imprinting disorder». Medicalxpress.com. Consultado em 18 de junho de 2015
↑ Runte, Maren; Varon, Raymonda; Horn, Denise; Horsthemke, Bernhard; Buiting, Karin (1 de fevereiro de 2005). «Exclusion of the C/D box snoRNA gene cluster HBII-52 from a major role in Prader–Willi syndrome». Human Genetics (em inglês). 116 (3): 228–230. ISSN 0340-6717. doi:10.1007/s00439-004-1219-2
↑ Skryabin BV, Gubar LV, Seeger B, Pfeiffer J, Handel S, Robeck T, Karpova E, Rozhdestvensky TS, Brosius J (2007). «Deletion of the MBII-85 snoRNA gene cluster in mice results in postnatal growth retardation». PLoS Genet. 3 (12): e235. PMC 2323313 . PMID 18166085. doi:10.1371/journal.pgen.0030235
. PMID 18166085. doi:10.1371/journal.pgen.0030235
↑ Sahoo T, del Gaudio D, German JR, Shinawi M, Peters SU, Person RE, Garnica A, Cheung SW, Beaudet AL (2008). «Prader-Willi phenotype caused by paternal deficiency for the HBII-85 C/D box small nucleolar RNA cluster.». Nat Genet. 40 (6): 719–21. PMC 2705197. PMID 18500341. doi:10.1038/ng.158
↑ Ding F, Li HH, Zhang S, Solomon NM, Camper SA, Cohen P, Francke U (2008). Akbarian, Schahram, ed. «SnoRNA Snord116 (Pwcr1/MBII-85) deletion causes growth deficiency and hyperphagia in mice». PLoS ONE. 3 (3): e1709. PMC 2248623. PMID 18320030. doi:10.1371/journal.pone.0001709 !CS1 manut: Nomes múltiplos: lista de autores (link)
↑ Ding F, Prints Y, Dhar MS, Johnson DK, Garnacho-Montero C, Nicholls RD, Francke U (2005). «Lack of Pwcr1/MBII-85 snoRNA is critical for neonatal lethality in Prader-Willi syndrome mouse models». Mamm Genome. 16 (6): 424–31. PMID 16075369. doi:10.1007/s00335-005-2460-2
↑ de Smith, Adam J.; Purmann, Carolin; Walters, Robin G.; Ellis, Richard J.; Holder, Susan E.; Van Haelst, Mieke M.; Brady, Angela F.; Fairbrother, Una L.; Dattani, Mehul (1 de setembro de 2009). «A deletion of the HBII-85 class of small nucleolar RNAs (snoRNAs) is associated with hyperphagia, obesity and hypogonadism». Human Molecular Genetics. 18 (17): 3257–3265. ISSN 1460-2083. PMID 19498035. doi:10.1093/hmg/ddp263
↑ ab Killeen, Anthony A. (2004). «Genetic Inheritance». Principles of Molecular Pathology. [S.l.]: Humana Press. p. 41. ISBN 978-1-58829-085-4
↑ Cassidy, Suzanne B.; Driscoll, Daniel J. (10 de setembro de 2008). «Prader–Willi syndrome». European Journal of Human Genetics (em inglês). 17 (1): 3–13. ISSN 1018-4813. PMID 18781185. doi:10.1038/ejhg.2008.165
↑ Davies, P. S.; Evans, S.; Broomhead, S.; Clough, H.; Day, J. M.; Laidlaw, A.; Barnes, N. D. (1 de maio de 1998). «Effect of growth hormone on height, weight, and body composition in Prader-Willi syndrome». Archives of Disease in Childhood. 78 (5): 474–476. ISSN 1468-2044. PMID 9659098
↑ Höybye, Charlotte; Hilding, Agneta; Jacobsson, Hans; Thorén, Marja (1 de maio de 2003). «Growth hormone treatment improves body composition in adults with Prader-Willi syndrome». Clinical Endocrinology. 58 (5): 653–661. ISSN 0300-0664. PMID 12699450
↑ «Double-Blind, Placebo Controlled, Phase 3 Trial of ZGN-440 (Beloranib) in Obese Subjects With Prader-Willi Syndrome – Full Text View – ClinicalTrials.gov». Consultado em 24 de Março de 2016
↑ «Potential PWS Treatments Currently in Development». Foundation for Prader-Willi Research. Consultado em 24 de Março de 2016
↑ «Unmet Needs in Prader-Willi Syndrome». Consultado em 24 de Março de 2016
↑ «An Efficacy, Safety, and Pharmacokinetics Study of Beloranib in Obese Subjects With Prader-Willi Syndrome». Consultado em 24 de março de 2016
↑ «Zafgen Announces Initial Results from Phase 2a Study of Beloranib in Patients with Prader-Willi Syndrome». Consultado em 24 de Março de 2016
↑ «UPDATE 4-Zafgen halts obesity drug trial after second patient death». Reuters. 3 de dezembro de 2015. Consultado em 26 de fevereiro de 2016
↑ Cassidy, Suzanne B.; Driscoll, Daniel J. (10 de setembro de 2008). «Prader–Willi syndrome». European Journal of Human Genetics (em inglês). 17 (1): 3–13. ISSN 1018-4813. PMID 18781185. doi:10.1038/ejhg.2008.165
↑ Tweed (Setembro de 2009). «Shawn Cooper Struggles with Prader Willi Syndrome». AOL Health
↑ Mary Jones. «Case Study: Cataplexy and SOREMPs Without Excessive Daytime Sleepiness in Prader Willi Syndrome. Is This the Beginning of Narcolepsy in a Five Year Old?». European Society of Sleep Technologists. Consultado em 6 de abril de 2009
↑ «Dog Eat Dog». Csifiles.com. Consultado em 12 de Junho de 2009
↑ «Can't Stop Eating». Channel4.com. 2006. Consultado em 12 de Junho de 2009
↑ «Extreme Makeover: Home Edition Articles on AOL TV». Aoltv.com. Consultado em 18 de junho de 2015
↑ Group urges more support for Prader-Willi sufferers, Taipei Times. Published December 24, 2011. Retrieved May 27, 2012.

