Imunoglobulina
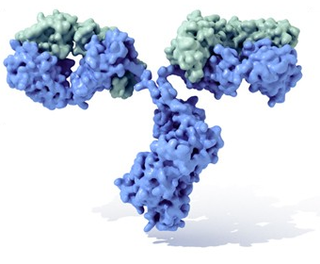
Molécula de imunoglobulina tipicamente com a sua forma em Y. Em azul observam-se as cadeias pesadas com quatro domínios Ig, enquanto que em verde mostram-se as cadeias leves. Entre o pé do Y (fracção constante, Fc) e os braços (Fab) existe uma parte mais fina conhecida como "região de dobradiça".
Os anticorpos (Ac) (também conhecidos como imunoglobulinas, abreviado Ig)[1] são glicoproteínas do tipo gamaglobulina, a fracção de globulinas mais abundante no plasma sanguíneo. Podem encontrar-se em forma solúvel no sangue ou noutros fluídos corporais dos vertebrados, ou podem estar inseridos na membrana plasmática, onde actuam como receptores nos linfócitos B e são empregues pelo sistema imunitário para neutralizar patógenos tais como bactérias patogénicas e viroses.[2] Em geral, considera-se que tanto anticorpo como imunoglobulina são termos equivalentes, sendo que o primeiro termo faz referência à função, enquanto que o segundo alude à estrutura. O termo gamaglobulina refere-se às propriedades electroforéticas das imunoglobulinas solúveis no soro sanguíneo, se bem que algumas imunoglobulinas migram com as fracções alfa, beta e inclusive com a albumina.
Um anticorpo é tipicamente constituído por unidades estruturais básicas, cada uma das quais com duas grandes cadeias pesadas e duas cadeias leves de menor peso molecular. A molécula de anticorpo tem forma de Y; as extremidades dos braços do Y são o fragmento Fab por onde se ligam ao antígeno; o pé do Y é o fragmento Fc. As moléculas dos anticorpos podem aparecer em separado, como monómeros, ou associarem-se entre si formando dímeros com duas unidades ou pentâmeros com cinco unidades. Os anticorpos são sintetizados por um tipo de leucócito denominado linfócito B ou célula B. Existem diferentes tipos de anticorpos, chamados isótipos, diferenciados pela forma da cadeia pesada que apresentem.São conhecidas cinco classes de isótipos em mamíferos que desempenham funções diferentes, contribuíndo para dirigir a resposta imunitária conforme cada tipo de corpo estranho que encontram, que são: IgA, IgD, IgE, IgG e IgM.[3]
Embora a estrutura geral de todos os anticorpos seja muito semelhante, uma pequena região do ápice da proteína é extremamente variável, permitindo a existência de milhões de anticorpos, cada um com uma extremidade ligeiramente diferente. Esta parte da proteína é conhecida como região hipervariável e dá lugar a milhões de anticorpos diferentes. Cada uma destas variantes pode ligar-se a um "alvo" diferente, que é o antígeno[4] Esta enorme diversidade de anticorpos permite ao sistema imunitário reconhecer uma diversidade igualmente elevada de antígenos. O anticorpo não reconhece o antígeno na sua globalidade, mas apenas reconhece certas partes dele. Essa parte do antígeno reconhecida pelo anticorpo denomina-se epítopo. Um antígeno pode ter múltiplos epítopos na sua superfície. Estes epítopos unem-se ao seu anticorpo numa interacção altamente específica que se denomina adaptação induzida, que permite aos anticorpos identificar e unir-se apenas ao seu único antígeno no meio dos milhões de moléculas diferentes que compõem um organismo.
O reconhecimento dum antígeno por um anticorpo deixa o antígeno marcado para ser atacado por outros componentes do sistema imunitário. Os anticorpos também podem neutralizar os seus objectivos directamente, mediante, por exemplo, a ligação a uma porção dum patógeno necessária para que este provoque uma infecção.
A extensa população de anticorpos e a sua diversidade é gerada por combinações ao acaso de um jogo de segmentos genéticos que codificam diferentes lugares de ligação ao antígeno (ou parátopos), que posteriormente, durante o desenvolvimento do linfócito, sofrem mutações aleatórias nesta zona do gene do anticorpo, o qual origina uma diversidade ainda maior.[3][5] Os genes dos anticorpos também se reorganizam num processo conhecido como comutação da classe das imunoglobulinas que troca a cadeia pesada por outra, criando um isótipo de anticorpo diferente mantendo, contudo, a região variável específica para o antígeno alvo. Isto possibilita que um só anticorpo possa ser usado pelas diferentes partes do sistema imunitário. A produção de anticorpos é a função principal do sistema imunitário humoral.[6]

A escultura Anjo do oeste (Angel of the West) (2008) de Julian Voss-Andreae basea-se na estrutura do anticorpo publicada por E. Padlan.[7] Desenhada para o campus Flórida do Instituto de Investigação Scripps,[8] o anticorpo está situado dentro dum anel que faz lembrar o Homem vitruviano de Leonardo da Vinci, destacando assim as dimensões similares do anticorpo e do corpo humano.[9]
Índice
1 História
2 Formas de anticorpos
2.1 Forma solúvel
2.2 Forma ancorada à membrana
3 Isótipos, alótipos e idiótipos
3.1 Isótipos
3.2 Alótipos
3.3 Idiótipo
4 Estrutura
4.1 Primeiros trabalhos
4.2 Domínios de imunoglobulina
4.3 Cadeia pesada
4.4 Cadeia leve
4.5 Regiões Fab e Fc
5 Função
5.1 Activação do complemento
5.2 Activação das células efetoras
5.3 Anticorpos naturais
6 Interacções antígeno-anticorpo
7 Diversidade das imunoglobulinas
7.1 Variabilidade de domínios
7.2 Recombinação V(D)J
7.3 Hipermutação somática e maturação da afinidade
7.4 Comutação de classe
7.5 Conversão genética
7.6 Fases finais da sintese de imunoglobulinas
7.7 Designações de especificidade
7.8 Anticorpos assimétricos
8 Evolução das imunoglobulinas
8.1 Animais pluricelulares
8.2 Deuteróstomos
8.3 Gnatóstomos
9 Aplicações médicas
9.1 Diagnóstico das doenças
9.2 Tratamentos terapêuticos
9.3 Terapia pré-natal
10 Aplicações em investigação científica
10.1 Obtenção
10.2 Aplicações
10.3 Registo e informação
11 Variantes de anticorpos em medicina e investigação
12 Previsão da estrutura
13 Ver também
14 Notas
15 Referências
História |


A primeira menção do termo "anticorpo" aparece num texto do bacteriologista alemão Paul Ehrlich (1854-1915). O termo "Antikörper" (a palavra alemã para anticorpo) aparece na conclusão de seu artigo "Estudos Experimentais sobre Imunidade", publicado em Outubro de 1891, que estabelece que "se duas substâncias dão origem a dois Antikörper diferentes, então elas próprias têm que ser obrigatoriamente diferentes".[10] No entanto, o termo não foi inicialmente aceito, tendo sido propostos vários termos alternativos para anticorpo; incluindo Immunkörper, Amboceptor, Zwischenkörper, substance sensibilisatrice, copula, Desmon, philocytase, fixateur e Immunisin.[10] A palavra anticorpo teria uma analogia formal com a palavra antitoxina e um conceito idêntico para Immunkörper (em português: corpo imune).[10] Como tal, a construção original da palavra contém um defeito lógico; antitoxina sugere algo que se opõe a uma toxina, enquanto que anticorpo sugere um corpo que se opõe a algo.[10]
Em 1890 foi quando se começou o estudo dos anticorpos, quando o fisiologista alemão Emil Adolf von Behring (1854-1917) e o bactereologista japonês Shibasaburo Kitasato (1852-1931), descreveram em 1890, pela primeira vez, as actividades dos anticorpos contra as toxinas da difteria e do tétano.[11] Behring e Kitasato propuseram a teoria da imunidade humoral, que estabelecia a existência dum mediador no soro sanguíneo que poderia reagir com um antígeno estranho, dando-lhe o nome de anticorpo.[12][13] A sua ideia fez com que, em 1897, Paul Ehrlich propusesse a teoria da cadeia lateral sobre a interacção entre o antígeno e anticorpo e elaborasse a hipótese de que existiam receptores (descritos como "cadeias laterais") na superfície das células que se poderiam unir especificamente a toxinas — numa interacção de tipo chave-fechadura— e que esta reacção de acoplamento seria o disparo para a produção de anticorpos.[14]
Em 1904, seguindo a ideia de outros investigadores de que os anticorpos se encontravam livres no sangue, Almroth Wright sugeriu que os anticorpos solúveis recobriam as bactérias para assinalá-las para a sua fagocitose e destruição num processo denominado opsonização.[15]


Na década de 1920, o imunologista americano Michael Heidelberger (1888-1991) e o médico canadense Oswald Avery (1877-1955) descobriram a natureza dos anticorpos postulados ao observar que os antígenos podiam ser precipitados por anticorpos, o que os levou à demonstração de que os anticorpos eram compostos protéicos.[16]
Desta forma, começava uma linha de pesquisa voltada para a elucidação das propriedades bioquímicas da interação antígeno-anticorpo, como a análise detalhada da ligação de um antígeno com o seu anticorpo, realizada pelo médico americano John Marrack (1886–1976) em finais da década de 1930.[17] Logo depois, na década de 1940, deu-se o principal avanço, quando Linus Pauling confirmou a teoria da chave-fechadura proposta por Ehrlich mostrando que as interacções entre anticorpos e antígenos dependiam mais da sua forma do que da sua composição química.[18] Em 1948, Astrid Fagreaus descobriu que os linfócitos B transformados em células plasmáticas eram responsáveis pela produção de anticorpos.[19]

A Universidade Rockefeller, antigo Instituto de Medicina, onde se desenvolveram boa parte dos avanços no estudo dos anticorpos.
Os trabalhos de investigação que se seguiram concentraram-se na caracterização da estrutura molecular dos anticorpos:
No começo da década de 1960 produziu-se o principal avanço neste sentido, com a descoberta por Gerald M. Edelman e Joseph Gally da cadeia leve,[20] e a compreensão de que esta era idêntica à proteína de Bence Jones descrita em 1845 por Henry Bence Jones.[21] Edelman continuou com a descoberta de que os anticorpos estavam compostos por cadeias leves e pesadas unidas por ligações dissulfeto. Na mesma época, Rodney Porter caracterizou as regiões de ligação do antígeno no anticorpo (Fab ou F antigen-binding) e a cola do anticorpo (Fc) no tipo IgG.[22] Conjuntamente, estes cientistas deduziram a estrutura e a sequência completa de aminoácidos da IgG, pelo qual receberam ex aequo o Prémio Nobel de Fisiologia ou Medicina em 1972.[22]
Apesar de a maioria destes primeiros estudos se centrarem nas IgM e IgG, identificaram-se também outros isótipos de imunoglobulinas na década de 1960: Thomas Tomasi descobriu os anticorpos secretados (IgA)[23] e David S. Rowe e John L. Fahey identificaram a IgD,[24] e a IgE foi identificada por Kikishige Ishizaka e Teruki Ishizaka como uma classe de anticorpos envolvidos nas reacções alérgicas.[25] Em 1975 César Milstein e Georges J.F. Köhler idealizaram o método para a produção de anticorpos monoclonais.[26] Em 1976, os estudos genéticos revelaram a base da vasta diversidade dos anticorpos quando Susumu Tonegawa identificou a recombinação somática dos genes de imunoglobulina.[27]
Formas de anticorpos |
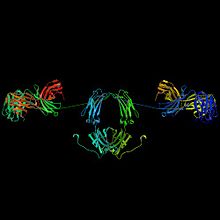
Diagrama de fitas da estrutura molecular de uma imunoglobulina A, um tipo de Ig segregável.
Os linfócitos B activados se diferenciam em células plasmáticas, cuja função é a produção de anticorpos solúveis ou ainda em linfócitos B de memória, que sobrevivem no organismo durante os anos seguintes para possibilitar que o sistema imunitário se lembre do antígeno e responda mais rápido a futuras exposições ao agente imunógeno.[28] Os anticorpos são, portanto, um produto essencial do sistema imunitário adaptativo que aprendem e lembram-se das respostas a patógenos invasores. Os anticorpos encontram-se em duas formas: na forma solúvel segregada no sangue e noutros fluídos do corpo e na forma unida à membrana celular que está ancorada à superfície dum linfócito B.
Forma solúvel |
Os anticorpos solúveis são segregados por um linfócito B activado (na sua forma de célula plasmática) para unir-se a substâncias estranhas e sinalizá-las para a sua destruição pelo resto do sistema imunitário. Também se lhes poderia chamar anticorpos livres até que se unam a um antígeno e acabem como parte dum complexo antígeno-anticorpo ou denominá-los anticorpos segregados.
Nesta forma solúvel as imunoglobulinas unem-se a moléculas adicionais. Nas IgM, por exemplo, encontramos uma glicoproteína unida à fracção constante através de pontes dissulfeto com cerca de 15 KDa, chamada cadeia J. Ao isotipo IgA une-se-lhe a chamada "peça de secreção". Trata-se de uma glicoproteína que se forma nas células epiteliais e glândulas exócrinas e que, posteriormente, se une à imunoglobulina para facilitar a sua secreção.[29]
Forma ancorada à membrana |
A forma ancorada à membrana de um anticorpo poder-se-ia chamar imunoglobulina de superfície (sIg) ou imunoglobulina de membrana (mIg), uma vez que não é segregado: sempre está associado à membrana plasmática. Faz parte do receptor do linfócito B (BCR), que permite que este detecte quando um antígeno específico está presente no organismo, desencadeando a activação do linfócito B.[30] O BCR é composto por anticorpos IgD ou IgM ligados à superfície de membrana e aos seus heterodímeros associados Ig-α e Ig-β que têm capacidade de realizar a transdução de sinais do reconhecimento do anticorpo para o interior da célula.[31] Um linfócito B humano comum tem entre 50 000 e 100 000 anticorpos unidos à sua superfície.[31] Após o acoplamento do antígeno, estes agrupam-se formando grandes adesivos cujo diâmetro pode ser superior a 1μm em balsas lipídicas que isolam os BCR (receptores da célula B) da maior parte dos restantes receptores de sinalização celular.[31] Estes adesivos poderiam melhorar a eficiência da resposta imune celular.[32] Nos seres humanos, a superfície celular encontra-se livre de outras proteínas ao redor dos receptores dos linfócitos B em distâncias com alguns milhares de ångströms,[31] o que reduz de tal maneira as influências que competem com a sua função, que pode-se dizer que isola os BCR.
Isótipos, alótipos e idiótipos |
Isótipos |
Os anticorpos podem existir em diferentes formas conhecidas como isótipos ou classes. Nos mamíferos existem cinco isótipos diferentes de anticorpos, conhecidos como IgA, IgD, IgE,IgG e IgM com diferentes cadeias pesadas. Possuem o prefixo "Ig" que significa imunoglobulina, e diferenciam-se pelas suas propriedades biológicas, localizações funcionais e habilidade para lidar com diferentes antígenos, como mostrado na tabela 1.[33]
| Nome | Tipos | Descrição | Complexos |
| IgA | 2 | Encontrado em áreas de mucosas, como os intestinos, trato respiratório e trato urogenital, prevenindo sua colonização por patógenos. É passado para o neonato via aleitamento.[34] |  |
| IgD | 1 | Funciona principalmente como uma receptor de antígeno nas células B virgens.[35] Suas funções são menos definidas do que as dos outros isotérmicos. | |
| IgE | 1 | Se liga a alérgenos e ativam os mastócitos - responsáveis pela liberação de histamina- e basófilos. (Reação de hipersensibilidade Inata) → alergia. Também protege contra parasitas helmintos.[6] | |
| IgG | 4 | Participa da opsonização; activação do sistema de complemento (inflamação e fagocitose); citotoxicidade mediada por células dependentes de anticorpo; Inibição por feedback das células B. Além de ser o único tipo de Ig que ultrapassa a barreira placentária.[6] | |
| IgM | 1 | Expressa na superfície das células B virgens. Elimina patógenos nos estágios iniciais da imunidade mediada pelas células B antes que haja IgG suficiente - activação do sistema de complemento.[6][35] |
O isótipo altera-se durante o desenvolvimento e a activação dos linfócitos B. Antes da maturação destes últimos, quando ainda não foram expostos ao seu antígeno, são conhecidos como linfócitos B virgens e só expressam o isótipo IgM na sua forma ancorada à superficie celular. Os linfócitos B começam a expressar tanto IgM como IgD ligadas à membrana quando alcançam a maturação e nesse momento estão prontos para responder ao seu antígeno.[36] A activação dos linfócitos B continua ao encontro e ligação deste com o seu antígeno, o que estimula a célula para que se divida e se diferencie numa célula produtora de anticorpos denominada plasmática. Nesta forma activada, os linfocitos B começam a segregar anticorpos em vez de ancorá-los à membrana. Algumas células filhas dos linfócitos B activados sofrem uma mudança isotípica, um mecanismo que faz com que a produção de anticorpos nas formas IgM ou IgD se transmute para os outros tipos, IgE, IgA ou IgG, que desempenham diferentes funções no sistema imunitário.
| Nome | Descrição |
|---|---|
| IgY | Encontra-se em aves e répteis; está relacionada com a IgG de mamíferos.[37] |
| IgW | Própria dos peixes elasmobranquios (tubarões, raias); relacionada com a IgD de mamíferos.[38] |
Alótipos |
Por alótipos entende-se os diferentes anticorpos que têm pequenas diferenças na sequência de aminoácidos na região constante das cadeias leves e pesadas produzidos pelos diferentes indivíduos de uma espécie, que se herdam de forma mendeliana.
Em seres humanos descreveram-se 3 tipos de determinantes alotípicos:
- Em 1956 Grubb e Laurell descobrem o sistema Gm na classe de imunoglobulinas IgG. Este sistema revelou os diversos alótipos das cadeias pesadas. Também permitiu diferenciar quatro subclasses nestas moléculas: IgG1, IgG2, IgG3 e IgG4 que estão determinadas geneticamente.[39]
- C. Ropartz e colaboradores descobriram em 1961 para o sistema Km (inicialmente chamado Inv), localizado na cadeia leve Kappa. Este alótipo está presente a todas as classes de imunoglobulina.
- Também existe o sistema ISf, situado na cadeia pesada γ1 da IgG1. A expressão desta especificidade aumenta com a idade, e dá-se em 25 % dos indivíduos antes dos 20 anos e em até 60 % depois dos 70 anos nos caucasóides.
- Os alótipos definidos pelo sistema Am situam-se nas IgA, e mais precisamente nas cadeias α2. Existem dois isótipos, α1 e α2, que caracterizam as subclasses Am1 e Am2 das IgA.[40]
Idiótipo |
O idiótipo consiste nas diferenças encontradas nas Ig na parte hipervariável das cadeias pesada e leve, que são próprias das moléculas de anticorpos pertencentes a um clone em particular. Este elemento faz parte ou está muito próximo ao sítio de reconhecimento do antígeno, e está situado na porção variável Fab. O parátopo é a parte da região variável do anticorpo que se une ao antígeno. Face aos idiótipos formar-se-iam anticorpos (anticorpos de anticorpos). Segundo a teoria de Jerne, a formação de anticorpos anti-idiótipo formaria uma rede (rede de Jerne) cuja função seria regular a síntese de novas imunoglobulinas.[29]
Estrutura |
Os anticorpos são proteínas plasmáticas globulares pesadas (~150 kDa), também conhecidas por imunoglobulinas. Têm cadeias de açúcares unidas a algum dos seus resíduos de aminoácidos;[41][42] por outras palavras, são glicoproteínas.[41] A unidade básica funcional dos anticorpos é o monómero de imunoglobulina, que contém uma só unidade de Ig. Os anticorpos segregados também podem ser dímeros com duas unidades de Ig, como no caso das IgA, tetraméricos com quatro unidades Ig como nas IgM de teleósteos, ou pentaméricos com cinco unidades de IgM, como no caso das IgM de mamíferos.[43]
Primeiros trabalhos |

As imunoglobulinas constam de diferentes domínios, que por sua vez se agrupam em duas cadeias pesadas (vermelho e azul na figura) e as duas cadeias leves (verde e amarelo) do anticorpo. Os domínios da imunoglobulina são compostos por entre 7 (no caso da IgC) e 9 (na IgV) pregas β.
As primeiras investigações sobre a estrutura dos anticorpos foram realizadas por meio de simples digestões com pepsina e papaína por Rodney Robert Porter e Gerald M. Edelman, seguidas de electroforese. Ambos receberam com isto o Prémio Nobel de medicina em 1972. Também foi importante a figura de Alfred Nisonoff:
- Na década de 1950, Porter fez uma digestão suave com papaína, obtendo três fragmentos, dois dos quais retinham a especificidade de antígeno (fragmento Fab, do inglês ab → antigen-binding), enquanto que o terceiro não mostrava actividade de união e podia cristalizar (fragmento Fc, c → cristalizável).
- Em 1959, Edelman, utilizando 2-mercaptoetanol e ureia, seguido de electroforese, conseguiu isolar as cadeias leves e pesadas, ao dissociar as suas ligações dissulfeto e não covalentes.
- Nesse mesmo ano, Porter identificou os componentes das cadeias leves e pesadas que se encontravam nos seus fragmentos digeridos com papaína e pepsina, e conseguiu medir os seus pesos moleculares.
- Em 1960, Nisonoff demonstrou que a digestão com pepsina de IgG produzia um fragmento bivalente, que na realidade é formado por outros dois, que ele denominou F (ab')2.[44]
Domínios de imunoglobulina |
O monómero de Ig é uma molécula em forma de "Y" que consiste de duas cadeias de polipéptido; duas cadeias pesadas idênticas e duas cadeias leves idênticas conectadas por ligações dissulfeto. Cada cadeia é composta por domínios estruturais chamados domínios Ig. Estes domínios contêm entre 70 e 110 aminoácidos e classificam-se em diferentes categorias, por exemplo em variáveis (IgV) e constantes (IgC) de acordo com o seu tamanho e função.[45] Têm uma "prega de imunoglobulina" característica no qual duas folhas beta geram uma forma de "sandwich", permanecendo juntas por interacções entre cisteínas bem conservadas ao longo da evolução, assim como outros aminoácidos carregados.
Cadeia pesada |
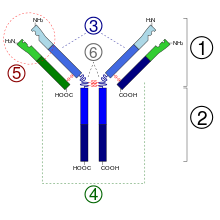
1. Região Fab.
2. Região Fc3.
3. Cadeia pesada com um domínio variável (VH) seguido por um domínio constante (CH1), uma região de dobradiça, e mais dois constantes, os domínios (CH2 e CH3).
4. Cadeia leve com um domínio variável (VL) e um constante (CL).
5. Lugar de união com o antígeno (parátopo)
6. Regiões de dobradiça.
Há cinco tipos de Ig em mamíferos que se designam por letras gregas: α, δ, ε, γ e μ.[4] O tipo de cadeia pesada presente define a classe (isótipo) do anticorpo. Estas cadeias encontram-se nos anticorpos IgA, IgD, IgE, IgG, e IgM, respectivamente. As diferentes cadeias pesadas diferem em tamanho e composição: α e γ contêm aproximadamente 450 aminoácidos, enquanto que μ e ε possuem aproximadamente 550 aminoácidos.[4]
As cadeias pesadas γ, α e δ possuem uma região constante composta por três domínios estruturais Ig em tandem e uma região de dobradiça para lhe proporcionar flexibilidade.[33] As cadeias pesadas μ e ε possuem uma região constante composta por quatro domínios de imunoglobulina[4] A região variável da cadeia pesada é diferente nos anticorpos produzidos nos diferentes linfócitos B, mas é idêntica para todos os anticorpos produzidos pelo mesmo linfócito B ou pela sua linha clonal. A região variável de cada cadeia pesada é de aproximadamente 110 aminoácidos e é composta por um único domínio Ig.
Recentemente pôde determinar-se a topologia in vivo do gene da cadeia pesada, Igh, tendo este sido um dos primeiros estudos nesse campo. O resultado é que a cromatina se dispõe formando circulos sucessivos unidos por linkers, dando lugar a formas parecidas com uma flor. A posição relativa dos diferentes segmentos varia drasticamente ao longo do desenvolvimento do linfócito B, permitindo assim um maior alcance de interacções genómicas.[46]
Cadeia leve |
Nos mamíferos existem dois tipos de cadeia leve, chamados lambda (λ) e kappa (κ).[4] Uma cadeia leve contém dois domínios sucessivos: um domínio constante e outro variável. O comprimento aproximado da cadeia leve é de 211 a 217 aminoácidos.[4] Cada anticorpo contém duas cadeias leves que são sempre idênticas. Só um tipo de cadeia leve, κ ou λ, encontra-se presente dentro do mesmo anticorpo em mamíferos. Outros tipos de cadeias leves como a cadeia iota (ι), encontram-se em vertebrados inferiores, como os peixes condrictios e teleósteos.
Regiões Fab e Fc |
Algumas partes do anticorpo têm funções únicas. As extremidades dos braços do "Y", por exemplo, contêm o lugar que se une ao antígeno e, portanto, reconhecem elementos estranhos específicos e neles esconde as extremidades N-terminal das cadeias polipeptídicas leve e pesada. Esta região do anticorpo chama-se fragmento de ligação do antígeno ou região Fab. É composta por um domínio constante e outro variável de cada uma das cadeias leve e pesada do anticorpo.[47] O parátopo é formado pelos domínios variáveis das cadeias pesada e leve na sua extremidade amino terminal. A função que a base ou pé do "Y" desempenha consiste em modular a actividade da célula imunitária. Esta região chama-se fragmento cristalizável ou Fc e é composta por dois ou três domínios constantes de ambas as cadeias pesadas, o tipo de cadeia pesada depende da classe do anticorpo. O pé termina nas extremidades C-terminal das cadeias pesadas.[4] Mediante a ligação a proteínas específicas a região Fc assegura que cada anticorpo gera uma resposta imunitária adaptada para um antígeno específico.[48] A região Fc também se une a vários receptores celulares como o receptor do Fc e outras moléculas do sistema imunitário como as proteínas do complemento. Ao efectuar isto, intervém na mediação de diferentes efeitos fisiológicos como o reconhecimento de partículas opsonizadas (unindo-se a FcγR), lise celular (unindo-se ao complemento) e desgranulação dos mastócitos, basófilos e eosinófilos (unindo-se a FcεR).[33][49]
Função |

Como os anticorpos se encontram na forma livre na corrente sanguínea, diz-se que fazem parte do sistema imunitário humoral. Os anticorpos circulantes são produzidos por linhas clonais de linfócitos B que respondem especificamente a um antígeno que pode ser um fragmento de proteína da cápside viral, por exemplo. Os anticorpos contribuem para a imunidade de três formas: podem impedir que os patógenos entrem nas células ou as danifiquem ao unir-se a elas (neutralização). Podem estimular a eliminação dum patógeno pelos macrófagos e outras células recubrindo o patógeno (opsonização) e podem desencadear a destruição directa do patógeno estimulando outras respostas imunes como a via do complemento (lise).[50]
Os principais tipos de acção dos anticorpos no sistema imunitário podem resumir-se a:
Neutralização, na qual anticorpos neutralizantes bloqueiam partes da superfície de uma célula bacteriana ou virião para fazer com que o seu ataque não seja efectivo ou neutralizam toxinas.
Aglutinação, na qual os anticorpos fazem com que as células estranhas "se peguem umas às outras" formando grupos compactos que são alvos atractivos para a fagocitose.
Precipitação, na qual os anticorpos fazem com que os antígenos solúveis no soro se "peguem entre si", o que provoca a sua precipitação fora da solução em grupos que depois serão fagocitados.
Activação do complemento (fixação), na qual os anticorpos que estão unidos à superfície das células estranhas favoreçam o ataque do complemento e a formação do complexo de ataque à membrana, o que conduz a lise das células estranhas e impulsiona a inflamação por atracção quimiotáctica de células inflamatórias.
Activação do complemento |
Os anticorpos que se unem à superfície dos antígenos, por exemplo numa bactéria, atraem os primeiros componentes da cascata do complemento por meio da sua região Fc e iniciam a activação do sistema "clássico" do complemento.[50] Isto provoca a morte da bactéria de duas maneiras:[6] Primeiro, a união das moléculas do complemento com o anticorpo marca o micróbio para a ingestão pelos fagócitos num processo chamado opsonização. Estes fagócitos são atraídos por determinadas moléculas do complemento. Em segundo lugar, alguns componentes do sistema do complemento formam um complexo de ataque à membrana para ajudar os anticorpos a matar a bactéria por meio da lise. Os anticorpos mais efectivos na activação do sistema do complemento são os do tipo IgM e os IgG subclasse 1 e 3 (IgG1 e IgG3).[51]
Activação das células efetoras |
Para combater os patógenos que se replicam no exterior das células, os anticorpos unem-se aos patógenos para ensamblá-los todos juntos provocando a sua aglutinação. Como um anticorpo tem pelo menos dois parátopos (os que são poliméricos têm mais) pode unir-se a mais de um antígeno acoplando-se a epítopos idênticos presentes nas superfícies desses antígenos. Ao cobrirem o patógeno, os anticorpos estimulam as funções efetoras contra este nas células que reconhecem a região Fc.[6]
Aquelas células que reconhecem os patógenos recobertos de anticorpos têm receptores do Fc que, como o próprio nome indica, interagem com a região Fc dos anticorpos IgA, IgG, e IgE. O acoplamento dum anticorpo específico com o receptor Fc de uma determinada célula desencadeia nela uma função efetora: os fagócitos realizarão a fagocitose, os mastócitos e os neutrófilos sofrerão a desgranulação, as células exterminadoras naturais libertarão citocinas e moléculas citotóxicas que finalmente acabarão por destruir o micróbio invasor. A activação das células exterminadoras naturais por anticorpos dá início a um mecanismo citotóxico conhecido como citotoxicidade mediada por células dependente de anticorpos (ADCC) - este processo pode explicar a eficácia dos anticorpos monoclonais usado em terapias biológicas contra cancro. Os receptores Fc são específicos do isótipo, o que dá uma maior flexibilidade ao sistema imunitário, afectando só o mecanismo imune adequado para os diferentes patógenos.[4]
Anticorpos naturais |
Os humanos e primatas superiores produzem também os chamados "anticorpos naturais" que estão presentes no soro sanguíneo antes que a infecção viral se origine. Os anticorpos naturais foram definidos como anticorpos que se produzem sem que exista uma infecção prévia, vacinação, exposição a outros antígenos alheios ou imunização passiva. Estes anticorpos podem activar a via clássica do complemento, o que origina a lise de partículas víricas com envoltura muito antes de activada a resposta imunitária adaptativa. Muitos anticorpos naturais são direccionados contra o dissacárido galactose α(1,3)-galactose (α-Gal), que se encontra como açucar terminal de proteínas da superfície celular glicosiladas, e são gerados em resposta à produção deste açucar pelas bactérias que se encontram no tracto digestivo humano.[52] Acredita-se que a rejeição de órgãos xenotransplantados se produza, em parte, como resultado de anticorpos naturais circulantes no soro do receptor que se unem a antígenos α-Gal expressados no tecido do doante.[53]
Interacções antígeno-anticorpo |
O parátopo do anticorpo (situado nas extremidades dos braços do Y) interage com o epítopo do antígeno. Um antígeno geralmente contém diferentes epítopos ao longo da sua superfície, dispostos de forma descontinua, e os epítopos dominantes dum antígeno são denominados determinantes antigénicos.
O antígeno e o anticorpo interagem por complementariedade espacial (como uma chave na sua fechadura). As forças moleculares implicadas nas interacções da extremidade Fab do anticorpo com o epítopo do antígeno são débeis e não específicas, como forças electrostáticas, ligações de hidrogénio interacções hidrofóbicas e forças de Van der Waals. Isto significa que a união entre o anticorpo e o antígeno é reversível, e a afinidade do anticorpo para o antígeno é relativa em vez de absoluta. Uma união relativamente fraca também significa que é possível que um anticorpo estabeleça reacções cruzadas com diferentes antígenos que têm diferentes afinidades relativas.
Frequentemente, assim que um anticorpo se une com o seu antígeno, estes formam um imunocomplexo, que funciona como um objecto unitário e podendo mesmo actuar como um antígeno do seu, que será contrariado por outros anticorpos. De modo similar, os haptenos são pequenas moléculas que não provocam uma resposta imunitária por sim sós, mas, assim que se unem a proteínas, o complexo resultante aduto hapteno-portador é antigénico.
Diversidade das imunoglobulinas |
Praticamente todos os microorganismos podem desencadear a resposta dos anti-anticorpos. O reconhecimento e erradicação bem sucedidos de tipos muitos distintos de micróbios requer que os anticorpos possuam uma enorme diversidade. A sua composição de aminoácidos varía para lhes permitir interagir com antígenos muito diferentes.[54] Estima-se que os seres humanos gerem cerca de 10 mil milhões de anticorpos diferentes, cada um dos quais com a capacidade de se unirem a um epítopo diferente.[55] Embora se gere um enorme repertório de diferentes anticorpos num mesmo indivíduo, o número de genes disponíveis para fabricar estas proteínas é limitado. Nos vertebrados evoluíram diferentes mecanismos genéticos complexos para permitir que os linfócitos B gerem esta diversidade a partir de um número relativamente pequeno de genes de anticorpos.[56]
Variabilidade de domínios |

As regiões hipervariáveis da cadeia pesada são mostradas em vermelho, PDB 1IGT.
A região (locus) do cromossoma que codifica um anticorpo é grande e contém vários genes diferentes para cada domínio de anticorpo, que se combinam entre si. O locus que contém os genes para cadeias pesadas (IGH @) pode ser encontrado em humanos no cromossoma 14 e loci contêm os genes lambda e kappa da cadeia leve (IGL@ e IGK@) são encontrados no 22 e no 2. Um destes domínios é conhecido como "domínio variável" e está presente em todas as cadeias leves e pesadas dos anticorpos, mas pode ser diferente entre os diferentes anticorpos gerados pelas diversas linhas de linfócitos B.
As diferenças entre os domínios variáveis estão localizadas em três bucles conhecidos como regiões hipervariáveis (HV-1, HV-2 e HV-3) ou regiões determinantes da complementaridade (CDR1, CDR2 e CDR3). As CDRs são mantidas dentro dos domínios variáveis por regiões de armação conservadas. O locus da cadeia pesada contém cerca de 65 genes diferentes de domínio variável, que diferem nos seus CDRs. Combinando estes genes com vários genes doutros domínios, gera-se um grande contingente de anticorpos com um alto grau de variabilidade. Esta combinação é chamada recombinação V(D)J.[57]
Recombinação V(D)J |

Esquema simples da recombinação V(D)J das cadeias pesadas de imunoglobulina.
A recombinação somática das imunoglobulinas, chamadas recombinação V(D)J, consiste na criação de uma região variável de imunoglobulina exclusiva formada pela combinação de vários segmentos genéticos (subgenes). A região variável de cada imunoglobulina pesada é codificada por várias partes, denominadas segmentos, chamados segmento variável (V), de diversidade (D) e de acoplamento — joining, em inglês— (J).[56] Os segmentos V, D e J encontram-se nas cadeias pesadas. Nas leves só encontramos os segmentos V e J. Há múltiplas cópias de todos estes segmentos organizadas em tandem no genoma dos mamíferos. Na medula óssea cada linfócito B em desenvolvimento ensambla a região variável da sua imunoglobulina selecionando e combinando um segmento V com um D e outro J na cadeia pesada e um V e outro J na cadeia leve. Como existem múltiplas cópias ligeiramente distintas para cada sequência genética dos segmentos, vão-se dar diferentes combinações que por intermédio deste processo geram um elevado número de parátopos e também diferentes especificidades de antígeno.[3] Curiosamente, o rearranjo de vários subgenes (isto é, a família V2) para a imunoglobulina de cadeia leve lambda é acoplado com a activação do microRNA miR-650, que influencia ainda mais a biologia dos linfócitos B.[58]
As proteínas RAG desempenham um importante papel na recombinação V(D)J ao cortarem o ADN em determinadas regiões.[59] Sem a presença destas proteínas a recombinação V(D)J não pode ocorrer.[59]
Após a produção de uma imunoglobulina funcional por um linfócito B durante a recombinação V(D)J este não poderá expressar nenhuma região variável diferente (este processo chama-se exclusão alélica). Portanto, cada linfócito B só pode produzir anticorpos que contenham um único tipo de cadeia variável.[4][60]
Hipermutação somática e maturação da afinidade |
Outro mecanismo que gera diversidade nos anticorpos tem lugar nos linfócitos B maduros. Após a activação pelo antígeno, os linfócitos B començam a proliferar rapidamente. Nestas células em rápida divisão, os genes que codificam os domínios variáveis das cadeias pesadas e leves sofrem uma grande taxa de mutação pontual através de um processo chamado hipermutação somática. Esta produz aproximadamente a troca dum nucleótido por um gene variável e célula em cada divisão celular.[5] Como consequência, qalquer célula filha de uma linha de linfócitos B adquire uma ligeira diferença na sequência de aminoácidos dos domínios variáveis das suas cadeias de anticorpos.
A hipermutação somática serve para aumentar a diversidade do reservatório de anticorpos e influi na afinidade da ligação entre o antígeno e o anticorpo.[61] Algumas mutações pontuais acabam por produzir anticorpos que têm interações mais fracas (baixa afinidade) com o seu antígeno do que o anticorpo original, enquanto que outras geram anticorpos com uma interação mais forte (alta afinidade).[62] Os linfócitos B que expressam anticorpos de elevada afinidade na sua superfície recebem um forte sinal para que sobrevivam durante as interações com outras células, enquanto que as que expressam anticorpos de baixa afinidade morrerão por apoptose.[62] Assim, os linfócitos B que expressam anticorpos com uma afinidade mais elevada pelo seu antígeno competirão com vantagem contra aqueles de menor afinidade na sua função e sobrevivência permitindo o aumento do nivel médio de afinidade dos anticorpos ao longo do tempo. O processo de produção de anticorpos com afinidade aumentada progressivamente denomina-se maturação da afinidade. A maturação da afinidade tem lugar nos linfócitos B maduros após a recombinação V(D)J e depende do suporte que recebam dos linfócitos T colaboradores.[63]
Comutação de classe |

Mecanismo de recombinação na troca de classe que permite a alteração do isótipo nos linfócitos B activados.
A comutação da classe das imunoglobulinas é um processo biológico que se dá após a activação dos linfócitos B, que permite a produção de diferentes classes de anticorpos (IgA, IgE, ou IgG).[3] Estas classes estão definidas pelas regiões constantes (C) da cadeia pesada da imunoglobulina. Inicialmente os linfócitos B virgens expressam só IgM e IgD na superfície celular (inseridas na membrana) com regiões de união ao antígeno idênticas. Cada isótipo está adaptado para uma função diferente e, portanto, depois da activação, é preciso um anticorpo com um mecanismo efetor IgG, IgA ou IgE para a eficaz eliminação do antígeno. A comutação da classe permite a descendência dum só linfócito B produzir anticorpos de diferentes isótipos. Apenas a região constante da cadeia pesada do anticorpo muda durante a troca da classe. As regiões variáveis, e, portanto, a especificidade do antígeno, permanece invariável. Deste modo, produzem-se efetores com a função adaptada para cada ameaça do antígeno. A troca de classe inicia-se por influência de citocinas. O isótipo gerado depende das citocinas presentes ao redor do linfócito B.[64]
O processo dá-se no gene da cadeia pesada por um mecanismo chamado à recombinação da troca de classe (ou CSR, do inglês class switch recombination). Este mecanismo baseia-se em sequências de nucleótidos conservadas, chamadas regiões de troca ou comutação (regiões S ou switch), que se encontram num ponto da sequência do ADN anterior aos genes da região constante (excepto na cadeia δ). A fibra de ADN divide-se pela actividade de determinadas enzimas em duas regiões S concretas.[65][66] O exão do domínio variável volta a emendar-se através dum processo chamado união de extremidade não-homóloga (ou NHEJ, non-homologous end joining) à região constante escolhida (γ, α ou ε). Este processo acaba formando um gene de imunoglobulina que codifica um anticorpo de um isótipo diferente.[67]
Conversão genética |
A conversão genética é uma troca não recíproca, na qual a sequência de ADN doadora não se modifica, enquanto que o gene aceptor adquire um segmento do doador por recombinação homóloga. Embora este mecanismo para gerar diversidade nos anticorpos seja conhecido há já algum tempo, nunca se lhe deu a devida importância até agora. Sabe-se que é muito importante em aves, as quais usam nas suas cadeias leves e pesadas um grande número de pseudogenes semelhantes às sequências D, situadas no princípio da sequência do gene das cadeias de imunoglobulina. Posteriormente, estes segmentos mudam somaticamente a única região V, e podem também estar submetidas à hipermutação.[68] Este mecanismo, curiosamente, também está presente em alguns outros mamíferos, como os coelhos.[69]
Fases finais da sintese de imunoglobulinas |
Uma vez reagrupados todos os segmentos, produz-se um só ARN mensageiro, que se poliadenila. Este ARN abandona o núcleo, dirigindo-se aos ribossomas do retículo endoplasmático rugoso, onde começa a sua tradução. Posteriormente produz-se a glicosilação dos anticorpos na parte luminal do retículo endoplasmático rugoso e a ensamblagem, cujo processo é o seguinte: H+H → H2+L → H2L2. Constitui uma excepção à IgM, na qual se une primeiro uma cadeia pesada com uma leve. O seu destino final é servir como receptor ou ser segregado, e depende de se possui ou não um fragmento adicional de 19 aminoácidos na extremidade C-terminal. Este péptido incorpora-se à síntese mediante um processo de emparelhamento ou splicing. A sua presença determina se possui ou não uma região hidrófoba que possa ancorar-se à membrana plasmática.[29]
Designações de especificidade |
Um anticorpo pode denominar-se mono-específico se tiver especificidade pelo mesmo antígeno ou epitopo,[70] ou bi-específico se tiver afinidade por dois antígenos diferentes ou dois epitopos distintos do mesmo antígeno.[71] Um grupo de anticorpos pode denominar-se polivalente (ou inespecífico) se tiver afinidade por varios antígenos[72] ou micro-organismos.[72] A chamada imunoglobulina intravenosa, se nada indica o contrário, consiste numa variedade de distintas IgG (IgG policlonal). Em contraste, os anticorpos monoclonais são anticorpos idênticos produzidos por uma só célula B.
Anticorpos assimétricos |
Os anticorpos heterodímeros, que são também assimétricos, possibilitam uma maior flexibilidade e novos formatos que permitam unir diversos fármacos aos braços do anticorpo. Um dos formatos gerais para o anticorpo heterodímero é o formato "protuberâncias-em-buracos" ("knobs-into-holes"). Este formato é específico da parte da cadeia pesada da região constante dum anticorpo. A parte das "protuberâncias" (“knobs”) é construída pela engenharia substituindo um aminoácido pequeno por outro maior. Este encaixa dentro do "buraco" ("hole"), o qual se constrói através da engenharia substituindo um aminoácido grande por outro mais pequeno. O que conecta as “protuberâncias” com os “buracos” são pontes dissulfeto entre as cadeias. A forma “protuberâncias-em-buracos” facilita a citotoxicidade mediada por células dependente do anticorpo. Os fragmentos variáveis da cadeia simples (scFv) encontram-se conectados ao domínio variável das cadeias pesada e leve por meio de um curto péptido de união ou linker. Este é rico em glicina, dando-lhe maior flexibilidade, e em serina/treonina, o que lhe confere a sua especificidade. Podem conectar-se dois fragmentos scFv distintos por meio da região de dobradiça ao domínio constante da cadeia pesada ou ao domínio constante da cadeia leve.[73] Isto resulta na bi-especificidade do anticorpo, permitindo-lhe interagir com as especificidades de ligação de dois antígenos diferentes.[74] O formato "protuberâncias-em-buracos" potencia a formação de heterodímeros, contudo não suprime a formação de homodímeros.
Para melhorarem ainda mais a função dos anticorpos heterodímeros, muitos cientistas estão a pensar produzir construções artificiais. Os anticorpos artificiais são motivos proteicos muito diversos que usam a estratégia funcional da molécula de anticorpo, não sendo porém limitados pelas restrições estruturais da armação e loops dos anticorpos naturais.[75] Poder controlar o desenho combinacional da sequência e o espaço tridimensional poderia trascender o desenho natural e permitir a união de diferentes combinações de medicamentos aos braços da molécula.
Os anticorpos heterodímeros possuem uma maior variedade de formas que podem adoptar e os fármacos que estão unidos aos braços não precisam ser os mesmos em todos os braços, o que permite fazer combinações diferentes de fármacos para utilizá-los no tratamento do cancro. Os farmacêuticos podem produzir anticorpos altamente funcionais bi-específicos e inclusive multi-específicos. O grau em que podem funcionar é impressionante dado que tal alteração da forma em relação à sua forma natural deveria, em princípio, levar a uma diminução da funcionalidade.
Evolução das imunoglobulinas |
O desenvolvimento de organismos complexos, com tecidos e várias linhas celulares necessitou do aparecimento de novas moléculas para assegurar, por um lado, que as células se aderiam a outras da mesma colónia e por outro, a defesa frente a possíveis intrusos parasitas ou patogénicos. Ao longo da evolução no desenvolvimento dos sistemas imunitários foram utilizadas três tipos de moléculas, as lectinas, as LLR e as imunoglobulinas. Os seus padrões operativos misturam-se por vezes para combinar as suas propriedades, embora existam poucas moléculas que contenham todas, como é o caso do gene da doença poliquística renal (PKD1).[76]
Muitos estudos integram provas importantes de que a superfamília das imunoglobulinas tem representantes entre as bactérias e arqueas ou que pelo menos as presentes neste grupo e as de eucariotas poderiam ter um antepassado comum, a partir do qual evoluíram separadamente. Assim, atribuíram-se a este grupo de proteínas bacterianas "semelhantes a imunoglobulinas" (BIg's) o receptor Fc de Ig em Streptococcus agalactiae, e a endoglucanase C de Cellumonas fimi.[77] Também existem outros exemplos como a invasina de Yersinia pseudotuberculosis ou as Lig (Leptospiral Ig-like, similares à Ig de Leptospira) de diversas espécies de Leptospira.[78][79] Após a sua descoberta em Streptococcus descobriu-se uma proteína deste tipo no fago T4. Nesta ocasião destacou-se que o seu papel estava relacionado com a adesividade celular.[80]
As proteínas com domínios Ig são comuns em eucariotas unicelulares, e até certo ponto a sua estrutura possui um carácter conservado.[81] Um exemplo disto seriam as alfa aglutininas de Saccharomyces cerevisiae. Trata-se de moléculas que medeiam a adesão celular e que guardam grandes parecenças com o grupo CD2-CD4 de humanos, cujo papel é em parte similar, intervindo neste último caso na adesão dos linfócitos T com as células que apresentam antígenos e células alvo.[82]
Animais pluricelulares |

Intermediários postulados na evolução molecular dos loci das Ig e dos TCR. Para uma explicação mais detalhada ver nota.[83]
Porém, é nos grupos de animais pluricelulares mais primitivos, os Parazoa, onde os cientistas tentam encontrar respostas à origem do sistema imunitário adaptativo. Neste sentido, têm-se feito vários trabalhos de investigação com este taxon, e em especial com uma esponja considerada como fóssil vivo, Geodia cydonium e também com Suberites domuncula. Na primeira podem-se encontrar muitos dos tipos de proteínas que também estão implicadas na imunidade dos mamíferos. Em especial, há dois tipos da superfamília das imunoglobulinas, as unidas ao receptor tirosína-quínase, e as moléculas não enzimáticas de adesão das esponjas. Curiosamente, os domínios correspondentes já demonstram polimorfismo, e embora cumpram funções que são simultaneamente de receptores e de moléculas de adesão celular, estão sobre-regulados em experimentos de enxertia.[84]
Em resumo, a superfamília das imunoglobulinas interveio no surgimento da multicelularidade ao manter a integridade estrutural dos organismos distinguindo o próprio do alheio. Isto ocorre graças à suas capacidades de gerar módulos, de unir-se especificamente a outras proteínas e de formar bastões, assim como de oligomerizar-se e gerar diversidade por splicing alternativo a partir de material genético limitado, convertem-se em modelos para mediar a aderência celular e como receptores de superfície de membrana.[85][86]
Na busca de precedentes do sistema imunitário adaptativo, encontramos vários exemplos de proteínas da superfamília das Ig em protóstomos que desempenham um papel na defesa imunitária, como a hemolina em bichos-da-seda, ou a proteína Dscam na Drosophila melanogaster, assim como proteínas relacionadas com o fibrinógeno com domínios Ig (FREP) em gastrópodes. Algumas destas proteínas, que representam uma barreira do tipo inato, podem ter isoformas solúveis e ancoradas à membrana, e gerar diversidade por splicing alternativo, e em zonas da molécula diferentes às cadeias variáveis de articulados.[87]
Deuteróstomos |
Muitos dos elementos do sistema imunitário adaptativo, incluindo as células especializadas, estão já pré-configurados nos organismos mais basais dos deuteróstomos. Foram feitas investigações no ouriço-do-mar Strongylocentrotus purpuratus, onde se encontrou um rico sistema imunitário com homólogos de importantes reguladores imunitários e hematopoiéticos de articulados, alguns deles críticos. Especula-se por isso que a pressão evolutiva chave para o desenvolvimento do complexo sistema imunitário em deuteróstomos não foi tanto a ameaça de patógenos como a existência de uma rica variedade de organismos simbiontes, dos quais é um bom exemplo a flora intestinal humana.[88] Como forma de ilustração, viu-se que 60% das espécies de equinodermos estão associados a simbiontes bacterianos.[89] Em tunicados continua o aumento da complexidade do sistema imunitário. Na ascídia Botryllus schlosseri, em experimentos com enxertos não compatíveis, foram detectadas muitas proteínas que revelam um complexo sistema imunitário inato e algumas proteínas com domínio de imunoglobulina,[90][91] e o mais surpreendente, é que também se pode encontrar um homólogo evidente de RAG1, contíguo a uma estrutura semelhante ao RAG2.[92] Porém, é em cefalocordados onde podemos encontrar os primeiros vestígios das nossas actuais imunoglobulinas. Realizaram-se muitos estudos no anfioxo Branchiostoma floridae, no qual se encontraram umas curiosas proteínas, chamadas VCBP (Proteínas tipo V que contêm domínios que se unem à quitina) com significativa homologia com as regiões V (variáveis) das imunoglobulinas, certamente implicadas na resposta imunitária, mas carentes da sua variabilidade. Estudos cristalográficos demonstraram que provavelmente se trata de uma molécula semelhante ao antepassado molecular das actuais regiões variáveis de articulados.[93][94][95]
Nos actuais ágnatos dão-se alguns dos traços que identificam um moderno sistema imunitário adaptativo, enquanto que noutros estão ausentes. Por um lado, existem células que já contêm grande parte da maquinaria molecular dos linfócitos. Isto sugere uma evolução deste tipo celular nos vertebrados mais basais, e possivelmente num protocordado. Existem várias proteínas Ig com domínios semelhantes a V, que inclusive contêm regiões V e J, ainda que estejam codificados num único exão e não sejam reorganizáveis. Porém, não possuem um sistema imunitário como o dos articulados, baseado nos antigos anticorpos solúveis, receptores de membrana, reorganização e união por RAG. Por sua vez, esta função é assumida por uma série de proteínas ricas em repetições de leucina, que inclusive podem sofrer uma complexa recombinação, como resultado da qual se obtém uma variabilidade equiparável à dos anticorpos (1014). Isto constitui um extraordinário exemplo de evolução paralela.[96]
Gnatóstomos |
Considera-se que o aparecimento do moderno sistema imunitário teve que ocorrer há 500 milhões de anos, durante a explosão câmbrica..[97] Provavelmente ocorreu num contexto em que existiriam muitas formas e combinações de módulos de proteínas das quais muitas desapareceriam pelas pressões selectivas. Neste sentido, uma das questões que suscita o apartado anterior é que se a evidência paleontológica indica que os peixes mandibulados actuais procedem dos ágnatos, e estes carecem do mesmo sistema de recombinação dos modernos sistemas imunitários, seguramente deveu existir um antepassado comum, um ostracodermo ancestral que possuísse os dois sistemas. De acordo com este ponto de vista, o sistema de recombinação V(D)J provavelmente representa um desenvolvimento evolutivo convergente numa ramificação dos ostracodermos que precedeu a linha dos gnatóstomos.[98][99]
Quanto às classes das imunoglobulinas, em peixes encontramos análogos da classe IgM, assim como da IgD, identificada em muitas espécies de teleósteos;[100] Também existem muitas classes exclusivas, como as que contêm as cadeias pesadas ζ e τ. Possivelmente são isótipos anteriores à IgM, evolutivamente falando.[101][102] No caso dos condríctios também encontramos isótipos exclusivos, para além da IgM. Trata-se das IgW (IgX ou IgNARC) e das IgNAR.[103]
O tipo IgG surge em anfíbios e segue em répteis, enquanto que o tipo IgA aparentemente surge num antepassado comum entre aves e mamíferos. O tipo IgE parece ser exclusivo de mamíferos.[29]
Aplicações médicas |
Diagnóstico das doenças |
Para vários diagnósticos é comum a detecção de anticorpos como prova de confirmação da patologia. Para isso realizam-se testes sorológicos.[104] Como exemplo, em ensaios bioquímicos para o diagnóstico de doenças,[105] estima-se o título de anticorpos contra o vírus de Epstein-Barr ou contra a doença de Lyme. Se esses anticorpos não forem encontrados significa que a pessoa não está infectada ou que o esteve há muito tempo e os linfócitos B que geravam estes anticorpos foram-se reduzindo de forma natural.
Na imunologia clínica analisa-se por nefelometria (ou turbidimetria) os níveis das diferentes classes de imunoglobulinas para caracterizar o perfil dos anticorpos do paciente.[106] Por exemplo, verificar uma elevação do título das diferentes classes de imunoglobulina pode ser útil, por vezes, para determinar a causa da lesão hepática a partir do diagnóstico diferencial.[1] Neste sentido, um título elevado de IgA indicaria cirrose alcoólica; se o que está elevado são as IgM suspeitar-se-ia que há uma hepatite viral e cirrose biliar primária, enquanto que as IgG apareceriam elevadas na hepatite vírica, auto-imune e cirrose.
As doenças auto-imunes podem ser diagnosticadas por anticorpos que se unem a epítopos do próprio organismo; muitos deles podem-se detectar através do exame ao sangue. Um exemplo seria o caso dos anticorpos direcionados contra os antígenos de superfície de eritrócitos na anemia hemolítica mediada pelo sistema imunitário, que se detectam a partir do teste de Coombs.[107] Esta comprovação também é utilizada para rastrear anticorpos na preparação de transfusões de sangue e também nas mulheres no período pré-natal.[107]
Na prática existem muitos métodos imunodiagnósticos baseados na detecção de complexos antígeno-anticorpo que se utilizam no diagnóstico de doenças infecciosas, por exemplo ELISA, imunofluorescência, western blot, imunodifusão e imunoelectroforese.
A nova química do dioxaborolano permite a marcação radioativa de flúor (18F) dos anticorpos, que possibilita o uso de imagens de tomografia por emissão de pósitrões (TEP ou PET) do cancro.[108]
Tratamentos terapêuticos |
A terapia de anticorpos monoclonais é utilizada no tratamento de doenças como a artrite reumatóide,[109]esclerose múltipla,[110]psoríase,[111] e muitas formas de cancro, incluíndo o linfoma não Hodgkin,[112]cancro colorrectal, cancro da cabeça e pescoço e cancro da mama.[113]
Algumas imunodeficiências, como a agamaglobulinemia ligada ao cromossoma X e a hipogamaglobulinemia consistem numa falta parcial ou completa de anticorpos.[114] Estas doenças são por vezes tratadas induzindo uma imunidade a curto prazo chamada imunidade passiva. Esta é adquirida por meio da infusão de anticorpos "pré-fabricados" em forma de soro sanguíneo humano ou animal, imunoglobulinas intravenosas ou anticorpos monoclonais no individuo afectado.[115]
Terapia pré-natal |
As chamadas imunoglobulinas Rho (D) ou imunoglobulinas anti-RhD são específicas do antígeno humano Rhesus D, também conhecido como fator Rhesus.[116]Destes anticorpos anti-RhD, são conhecidas várias marcas comerciais, tais como a RhoGAM, BayRHo-D, Gamulin Rh, HypRho-D, e WinRho SDF. O fator Rhesus é um antígeno que é encontrado nos eritrócitos. Indivíduos com Rhesus-positivos (Rh +) exibem este anticorpo na glicocálix dos seus eritrócitos, enquanto que os indivíduos (Rh-) não o possuem.
Durante o nascimento normal, o sangue fetal pode passar para a mãe devido a traumas no parto ou complicações na gravidez. No caso de incompatibilidade de Rh entre a mãe e o filho, a consequente mistura de sangue pode sensibilizar uma mãe Rh- contra o antígeno Rh do filho, fazendo com que nas gravidezes posteriores os fetos corram o risco de sofrer eritroblastose fetal.[117] Os Anti-RhD são administrados como parte do tratamento pré-natal para previr a sensibilização que possa vir a existir para assim evitá-la. Ao tratar a mãe com anticorpos anti-RhD antes e imediatamente após o trauma e o parto destrói-se o antígeno Rh do feto no corpo da mãe. Um aspecto importante é que isto acontece antes que o antígeno possa estimular os linfócitos B maternos que mais tarde poderiam "relembrar" o antígeno Rh gerando linfócitos B de memória. Portanto, o seu sistema humoral imunitário não fabricará anticorpos anti-Rh e não atacará os antígenos Rhesus do seu bebé actual ou futuro.[116]
Aplicações em investigação científica |

Imagem de imunofluorescência do citoesqueleto de eucariotas. Os filamentos de actina aparecem a vermelho, os microtúbulos em verde e o núcleo celular em azul.
Em investigação, os anticorpos purificados são utilizados em muitas aplicações. São muito habituais para identificar e localizar proteínas intra e extra-celulares. Os anticorpos são utilizados na citometria de fluxo, imunoprecipitação, em análises de western blot para identificar proteínas separadas por electroforese, e em imuno-histoquímica ou imunofluorescência. As proteínas também se podem detectar e quantificar com anticorpos utilizando as técnicas ELISA e ELISPOT.
Obtenção |
Os anticorpos específicos são produzidos em investigação injectando um antígeno num mamífero, como um rato, ratazana, coelho, cabra, ovelha ou cavalo para obter grandes quantidades de anticorpos. O sangue que se isola destes animais contém anticorpos policlonais (muitos anticorpos que se unem ao mesmo antígeno) no soro, o que se denomina anti-soro. Os antígenos também se podem injectar em frangos para a produção de anticorpos policlonais na gema do ovo.[118] Para obter anticorpos que sejam específicos para um só epítopo dum antígeno, ísolam-se os linfócitos que segregam anticorpos do animal e são imortalizados fundindo-os com uma linha de células cancerosas. As células fundidas são chamadas hibridomas, e crescem continuamente e segregam anticorpos em cultivo. As células de hibridoma simples são isoladas por clonagem por diluição para gerar clones celulares em que todos produzem o mesmo anticorpo; estes anticorpos são chamados anticorpos monoclonais.[119] Os anticorpos policlonais e monoclonais costumam purificar-se usando a proteína A/G ou cromatografia de afinidade a antígenos.[120]
Aplicações |
Em investigação, os anticorpos modificados são utilizados em muitas aplicações. Os anticorpos para aplicações de investigação podem encontrar-se directamente em fornecedores de anticorpos ou utilizando um motor de investigação especializado. Os anticorpos para investigação são essencialmente usados para identificar e localizar proteínas intra-celulares e extra-celulares. Os anticorpos são utilizados em citometria de fluxo para diferenciar os tipos celulares das proteínas que expressão; os diferentes tipos de células expressão diferentes combinações de moléculas de clusters de diferenciação (CD) na sua superfície, e produzem diferentes proteínas intra-celulares e segregáveis.[121] Também foram utilizadas em imunoprecipitação para separar proteínas e qualquera coisa unida a elas (co-imunoprecipitação) doutras moléculas num lisado celular,[122] em análises através da técnica western blot para identificar proteínas separadas por electroforese,[123] e em imuno-histoquímica ou imunofluorescência para examinar a expressão de proteínas em secções de tecidos ou para localizar proteínas dentro das células com o auxilio dum microscópio.[121][124] As proteínas podem também ser detectadas e quantificadas com anticorpos, usando as técnicas ELISA e ELISPOT.[125][126]
Registo e informação |
Os anticorpos utilizados em investigação são alguns dos reactivos mais poderosos e também problemáticos com um enorme número de factores que devem ser controlados em qualquer experimento como a reactividade cruzada, ou o reconhecimento por anticorpo de múltiplos epítopos e afinidades, o que pode variar amplamente dependendo das condições experimentais como o pH, solvente, estado do tecido etc. Foram feitas várias tentativas para melhorar tanto o método em que os investigadores validam os anticorpos[127][128] como os métodos em que informam dos anticorpos. Os investigadores que usam os anticorpos no seu trabalho precisam de registá-los correctamente para que a sua investigação possa ser reproduzida (e, portanto, comprovada, e qualificada por outros investigadores). Menos de metade dos anticorpos de investigação referenciados em artigos académicos podem ser identificados facilmente.[129] Os artigos publicados em F1000 em 2014 e 2015 proporcionam aos investigadores uma guia para indicação do uso de anticorpos em investigação.[130][131] O documento RRID, encontra-se co-publicado em 4 revistas que aplicam os padrões RRIDs para a citação de recursos de investigação, os quais utilizam dados de antibodyregistry.org como fonte de identificação de anticorpos.[132] (ver também o grupo em Force11[133])
Variantes de anticorpos em medicina e investigação |
Por vezes é necessário produzir anticorpos específicos. Para injectar um antígeno num mamífero, como um rato, ratazana ou coelho requer-se pouca quantidade; já numa cabra, ovelha ou cavalo são requeridas grandes quantidades. O sangue isolado destes animais contém anticorpos policlonais (múltiplos anticorpos que se unem ao mesmo antígeno) no soro sanguíneo, ao qual se denomina anti-soro. Também se podem injectar antígenos na gema de ovo de galinha para produzi-los.[134] Porém, para aplicações analíticas é preciso uma maior especificidade, sobretudo quando se trata de detectar moléculas muito pequenas, assim como quando se usam em aplicações terapêuticas nas quais se deseja bloquear ou detectar marcadores muito específicos. Por isso, a tecnologia dos anticorpos gerou algumas variantes de anticorpos, entre as quais destacam-se as seguintes:
- Anticorpos monoclonais
Se se desejar obter anticorpos específicos para um único epítopo de um antígeno, isolam-se linfócitos secretores de anticorpos dum animal e imortalizam-se fundindo-os com uma linha celular cancerígena. As células fundidas denominam-se hibridomas e continuarão a crescer e a segregar anticorpos no cultivo. Isolam-se as células de hibridoma individuais por meio da clonagem por diluição para gerar clones que produzam todos o mesmo anticorpo. A estes anticorpos dá-se-lhes o nome de anticorpos monoclonais.[135]
Os anticorpos mono e policlonais gerados podem-se purificar utilizando a proteína A/G ou cromatografia de afinidade ao antígeno.[136]
- Anticorpos da cadeia leve
É possível gerar artificialmente um anticorpo que conte apenas com as regiões variáveis da cadeia leve e pesada, unidas por um pequeno péptido ou um só aminoácido. Neste caso teremos anticorpos da cadeia leve ou scFv's. Actualmente aplicam-se em técnicas como a citometria de fluxo ou a imunohistoquímica.[137]
- Abzimas
A maioria dos anticorpos diferenciam-se doutras proteínas por não apresentarem catálise enzimática na sua função, pelo que tradicionalmente se consideram proteínas de reconhecimento de superfícies moleculares. Porém, na década de 1990 e princípios do século XXI, verificou-se em diversos estudos de imunologia anticorpos com propriedades catalíticas. Tais anticorpos receberam o nome de abzimas. É possível encontrá-los em baixas quantidades no soro de pessoas saudáveis. Um exemplo da existência das abzimas no corpo humano foi a detecção de abzimas contra o ADN no leite materno.[138] Entre outras, estas actividades catalíticas detectadas são a de peptidases inespecíficas e amilolíticas (degradação de amido). Por outro lado, observou-se um aumento no nível de abzimas em doenças auto-imunes. Porém, normalmente fabricam-se de forma artificial gerando anticorpos contra o composto intermediário de uma reacção para a qual se deseja criar uma enzima. Em certas ocasiões podem ter aplicações terapêuticas e industriais.[139][140]
- Nanoanticorpos
Existem propostas para a utilização terapêutica de anticorpos monoclonais de camélido, também chamados de nanocorpos ou nanoanticorpos. Estes constituem excepções no reino animal, dado o seu reduzido tamanho, por serem compostos unicamente por duas cadeias pesadas.[141] Tais peculiaridades permitir-lhes-iam aceder a localizações celulares e antígenos inacessíveis para os anticorpos normais, além de ser possível a sua administração oral.[142]
- Faboterápicos
Para obter antídotos contra venenos de picadas de animais como serpentes ou artrópodes, fabricam-se anti-soros a partir de soro cru ou altamente enriquecido em imunoglobulinas. Estes procedimentos produziam inicialmente um grande número de reacções alérgicas, como anafilaxias ou a doença do soro. Para evitá-lo, nos anos 40 e 50 realizaram-se estudos de proteólise para reduzir ao mínimo a parte da molécula envolvida na neutralização do veneno. Finalmente, constatou-se que o fragmento F(ab’)2, resultante da digestão com pepsina dos anticorpos, que carece das zonas efectoras da molécula, pode neutralizar igualmente os venenos. O professor Alejandro Alagón Cano propôs para este enfoque terapêutico o nome de faboterapia, observando-se uma incidência muito menor de reacções adversas ao soro, assim como um melhor alcance do compartimento extravascular.[143]
Previsão da estrutura |
A importância dos anticorpos na saúde e indústria biotecnológica demanda um conhecimento das suas estruturas em alta resolução. Esta informação é utilizada na engenharia de proteínas, modificando a afinidade de união aos antígenos, e identificando um epítopo dum determinado anticorpo. Um método comummente utilizado para determinar as estruturas dos anticorpos é a cristalografia de raios X. No entanto, cristalizar um anticorpo costuma ser trabalhoso e leva muito tempo. Os métodos computacionais proporcionam uma alternativa mais barata e rápida à cristalografia, mas os seus resultados são mais incertos, uma vez que não produzem estruturas empíricas. Os servidores web em linha como Web Antibody Modeling (WAM)[144] e Prediction of Immunoglobulin Structure (PIGS)[145] permitem fazer os modelos computacionais de regiões variáveis dos anticorpos. O Rosetta Antibody é um novo servidor para a previsão de estruturas da região FV de anticorpos, que incorpora técnicas sofisticadas para minimizar os bucles CDR e optimizar a orientação relativa das cadeias leve e pesada, assim como modelos de homologia que prediz a união bem-sucedida dos anticorpos com os seus antígenos específicos.[146]
A capacidade de descrever o anticorpo pela afinidade de união ao antígeno é suplementada pela informação sobre a estrutura de anticorpos e sequências de aminoácidos com o propósito de registar patentes.[147]
Existe uma variedade de métodos utilizados para sequenciação dum anticorpo, incluindo a degradação de Edman, ADNc, etc; apesar de um dos métodos modernos mais comuns para a identificação de peptídeos/proteínas seja a cromatografia líquida acoplada à espectrometria de massa em tandem (LC-MS/MS).[148] O alto volume de métodos de sequenciamento de anticorpos requer abordagens computacionais para a análise de dados, incluindo o sequenciamento "De Novo" de peptídeos directamente de espectros de massa em tandem[149] e métodos de pesquisa na base de dados que utilizam o armazenamento de sequências de proteínas.[150][151] Muitas versões de sequenciamento de proteína shotgun são capazes de aumentar a cobertura utilizando métodos de fragmentação CID/HCD/ETD[152] e outras técnicas, alcançando progressos significativos na tentativa de conseguir um sequenciamento total de proteínas, principalmente anticorpos. Outros métodos têm assumido a existência de proteínas similares,[153] uma conhecida sequência genómica, ou combinação de abordagens do "topo para a base".[154] As tecnologias actuais têm a capacidade de montar sequências de proteínas com elevada precisão, integrando o sequenciamento "De Novo" de peptídeos, intensidade, e resultados posicionais confiáveis a partir do banco de dados e pesquisas homólogas.[155]
Ver também |
- Anticorpo monoclonal
- Autoanticorpo
- Imunoensaio
- ELISA
Imunoglobulina A IgA
Imunoglobulina D IgD
Imunoglobulina E IgE
Imunoglobulina G IgG
Imunoglobulina M IgM
Notas |
- A maior parte do texto foi baseado na tradução do artigo «Antibody» na Wikipédia em inglês (acessado nesta versão).
Referências
↑ ab Rhoades RA, Pflanzer RG (2002). Human Physiology 4th ed. [S.l.]: Thomson Learning. p. 584. ISBN 0-534-42174-1
↑ Litman GW, Rast JP, Shamblott MJ; et al. (1993). «Phylogenetic diversification of immunoglobulin genes and the antibody repertoire». Mol. Biol. Evol. 10 (1): 60–72. PMID 8450761 !CS1 manut: Uso explícito de et al. (link) !CS1 manut: Nomes múltiplos: lista de autores (link)
↑ abcd Eleonora Market, F. Nina Papavasiliou (2003) V(D)J Recombination and the Evolution of the Adaptive Immune System PLoS Biology1(1): e16.doi:10.1371/journal.pbio.0000016
↑ abcdefghi Janeway CA, Jr; et al. (2001). Immunobiology. 5th ed. ed. [S.l.]: Garland Publishing. ISBN 0-8153-3642-X !CS1 manut: Uso explícito de et al. (link) !CS1 manut: Texto extra (link)
↑ ab Diaz M, Casali P (2002). «Somatic immunoglobulin hypermutation». Curr Opin Immunol. 14 (2): 235–40. PMID 11869898. doi:10.1016/S0952-7915(02)00327-8
↑ abcdef Pier GB, Lyczak JB, Wetzler LM (2004). Immunology, Infection, and Immunity. [S.l.]: ASM Press. ISBN 1-55581-246-5 !CS1 manut: Nomes múltiplos: lista de autores (link)
↑ Padlan, Eduardo (1994). «Anatomy of the antibody molecule». Mol. Immunol. 31 (3): 169–217. PMID 8114766. doi:10.1016/0161-5890(94)90001-9
↑ «New Sculpture Portraying Human Antibody as Protective Angel Installed on Scripps Florida Campus». Consultado em 12 de dezembro de 2008. Cópia arquivada em 18 de novembro de 2010
↑ «Protein sculpture inspired by Vitruvian Man». Consultado em 12 de dezembro de 2008. Cópia arquivada em 18 de novembro de 2010
↑ abcd Lindenmann, Jean (1984). «Origin of the Terms 'Antibody' and 'Antigen'». Scand. J. Immunol. 19 (4): 281–5. PMID 6374880. doi:10.1111/j.1365-3083.1984.tb00931.x. Arquivado do original em 18 de novembro de 2010
↑ Grundbacher FJ (13 de maio de 1992). «Behring's discovery of diphtheria and tetanus antitoxins.». Immunol Today. (5): 188-90. PMID 1642758. doi:10.1016/0167-5699(92)90125-Q
↑ «Emil von Behring — Biography». Consultado em 5 de junho de 2007. Cópia arquivada em 18 de novembro de 2010
↑ AGN (1931). «The Late Baron Shibasaburo Kitasato». Canadian Medical Association Journal. 25 (2): 206. PMC 382621 . PMID 20318414
. PMID 20318414
↑ Winau F, Westphal O, Winau R (2004). «Paul Ehrlich—in search of the magic bullet». Microbes Infect. 6 (8): 786–789. PMID 15207826. doi:10.1016/j.micinf.2004.04.003
↑ Silverstein AM (2003). «Cellular versus humoral immunology: a century-long dispute». Nat. Immunol. 4 (5): 425–428. PMID 12719732. doi:10.1038/ni0503-425
↑ Van Epps HL (2006). «Michael Heidelberger and the demystification of antibodies» (PDF). J. Exp. Med. 203 (1): 5. PMC 2118068. PMID 16523537. doi:10.1084/jem.2031fta. Cópia arquivada (PDF) em 18 de novembro de 2010
↑ Marrack, JR (1938). Chemistry of antigens and antibodies 2nd ed. London: His Majesty's Stationery Office. OCLC 3220539
↑ «The Linus Pauling Papers: How Antibodies and Enzymes Work». Consultado em 5 de junho de 2007. Cópia arquivada em 18 de novembro de 2010
↑ Silverstein AM (2004). «Labeled antigens and antibodies: the evolution of magic markers and magic bullets» (PDF). Nat. Immunol. 5 (12): 1211–1217. PMID 15549122. doi:10.1038/ni1140. Arquivado do original (PDF) em 18 de dezembro de 2009
↑ Edelman GM, Gally JA (1962). «The nature of Bence-Jones proteins. Chemical similarities to polypetide chains of myeloma globulins and normal gamma-globulins». J. Exp. Med. 116 (2): 207–227. PMC 2137388. PMID 13889153. doi:10.1084/jem.116.2.207
↑ Stevens FJ, Solomon A, Schiffer M (1991). «Bence Jones proteins: a powerful tool for the fundamental study of protein chemistry and pathophysiology». Biochemistry. 30 (28): 6803–6805. PMID 2069946. doi:10.1021/bi00242a001
↑ ab Raju TN (1999). «The Nobel chronicles. 1972: Gerald M Edelman (b 1929) and Rodney R Porter (1917–85)». Lancet. 354 (9183): 1040. PMID 10501404. doi:10.1016/S0140-6736(05)76658-7
↑ Tomasi TB (1992). «The discovery of secretory IgA and the mucosal immune system». Immunol. Today. 13 (10): 416–418. PMID 1343085. doi:10.1016/0167-5699(92)90093-M
↑ Preud'homme JL; Petit I; Barra A; Morel F; Lecron JC; Lelièvre E (2000). «Structural and functional properties of membrane and secreted IgD». Mol. Immunol. 37 (15): 871–887. PMID 11282392. doi:10.1016/S0161-5890(01)00006-2
↑ Johansson SG (2006). «The discovery of immunoglobulin E». Allergy and Asthma Proceedings. 27 (2 Suppl 1): S3–6. PMID 16722325
↑ Raju, T N (2000). «The Nobel chronicles. 1984: Niels Kai Jerne, (1911-94); César Milstein (b 1926); and Georges Jean Franz Köhler (1946-95)». The Lancet. 355 (9197). 75 páginas. PMID 10615922. doi:10.1016/S0140-6736(05)72025-0
↑ Hozumi N, Tonegawa S (1976). «Evidence for somatic rearrangement of immunoglobulin genes coding for variable and constant regions». Proc. Natl. Acad. Sci. U.S.A. 73 (10): 3628–3632. PMC 431171. PMID 824647. doi:10.1073/pnas.73.10.3628
↑ Borghesi L, Milcarek C (2006). «From B cell to plasma cell: regulation of V(D)J recombination and antibody secretion». Immunol Res. 36 (1-3): 27–32. PMID 17337763. doi:10.1385/IR:36:1:27
↑ abcd Peña Martínez, J (Coordinador) (1998). Imunologia. [S.l.]: Pirámide. ISBN 84-368-1213-1 Disponivel uma versão online em http://www.uco.es
↑ Parker D (1993). «T cell-dependent B cell activation». Annu. Rev. Immunol. 11 (1): 331–360. PMID 8476565. doi:10.1146/annurev.iy.11.040193.001555
↑ abcd Wintrobe, Maxwell Myer (2004). John G. Greer; John Foerster; John N Lukens; George M Rodgers; Frixos Paraskevas, eds. Wintrobe's clinical hematology 11 ed. Hagerstown, MD: Lippincott Williams & Wilkins. pp. 453–456. ISBN 978-0-7817-3650-3
↑ Tolar P, Sohn HW, Pierce SK (Fevereiro de 2008). «Viewing the antigen-induced initiation of B-cell activation in living cells». Immunol. Rev. 221 (1): 64–76. PMID 18275475. doi:10.1111/j.1600-065X.2008.00583.x
↑ abc Woof J, Burton D (2004). «Human antibody-Fc receptor interactions illuminated by crystal structures». Nat Rev Immunol. 4 (2): 89–99. PMID 15040582. doi:10.1038/nri1266
↑ Underdown B, Schiff J (1986). «Immunoglobulin A: strategic defense initiative at the mucosal surface». Annu Rev Immunol. 4: 389-417. PMID 3518747
↑ ab Geisberger R, Lamers M, Achatz G (2006). «The riddle of the dual expression of IgM and IgD». Immunology. 118 (4): 429-37. PMID 16895553 !CS1 manut: Nomes múltiplos: lista de autores (link)
↑ Goding J. «Allotypes of IgM and IgD receptors in the mouse: a probe for lymphocyte differentiation». Contemp Top Immunobiol. 8: 203–43. PMID 357078
↑ Lundqvist, ML; Middleton, DL; Radford, C; Magor, KE (2006). «Immunoglobulins of the non-galliform birds: antibody expression and repertoire in the duck». Dev Comp Immunol. 30 (1): 93–100. PMC 1317265. PMID 16150486. doi:10.1016/j.dci.2005.06.019
↑ Berstein, RM; Schluter, SF; Shen, S; Marchalonis, JJ (16 de abril de 1996). «A new high molecular weight immunoglobulin class from the carcharhine shark: implications for the properties of the primordial immunoglobulin.». Proc Natl Acad Sci U S A. 93 (8): 3289–3293. PMC 39599. PMID 8622930. doi:10.1073/pnas.93.8.3289
↑ Grubb, R., and Laurell, A. B., Acta Path. Microb. Scand., 39, 390 (1956). PMID 13381487
↑ Janeway, CA; Staff, VV. Tradução de Eva Sanz (2003). Inmunobiología: el sistema inmunitario en condiciones de salud y enfermedad. [S.l.]: Elsevier España. ISBN 978-84-458-1176-4 !CS1 manut: Nomes múltiplos: lista de autores (link)
↑ ab Maverakis E, Kim K, Shimoda M, Gershwin M, Patel F, Wilken R, Raychaudhuri S, Ruhaak LR, Lebrilla CB (2015). «Glycans in the immune system and The Altered Glycan Theory of Autoimmunity». J Autoimmun. 57 (6): 1–13. PMC 4340844. PMID 25578468. doi:10.1016/j.jaut.2014.12.002
↑ Mattu T, Pleass R, Willis A, Kilian M, Wormald M, Lellouch A, Rudd P, Woof J, Dwek R (1998). «The glycosylation and structure of human serum IgA1, Fab, and Fc regions and the role of N-glycosylation on Fc alpha receptor interactions». J Biol Chem. 273 (4): 2260–2272. PMID 9442070. doi:10.1074/jbc.273.4.2260
↑ Roux K (1999). «Immunoglobulin structure and function as revealed by electron microscopy». Int Arch Allergy Immunol. 120 (2): 85–99. PMID 10545762. doi:10.1159/000024226
↑ Stevenson, JR (18 de agosto de 2008). «Immunoglobulin Structure and Function». CAS, Universidade de Miami. Consultado em 25 de agosto de 2008. Cópia arquivada em 25 de setembro de 2008
↑ Barclay A (2003). «Membrane proteins with immunoglobulin-like domains – a master superfamily of interaction molecules». Semin Immunol. 15 (4): 215–223. PMID 14690046. doi:10.1016/S1044-5323(03)00047-2
↑ Murre C, e outros: (2008). «The 3D structure of the immunoglobulin heavy-chain locus: implications for long-range genomic interactions». Cell. 133 (2). PMID 18423198
↑ Putnam FW, Liu YS, Low TL (1979). «Primary structure of a human IgA1 immunoglobulin. IV. Streptococcal IgA1 protease, digestion, Fab and Fc fragments, and the complete amino acid sequence of the alpha 1 heavy chain». J Biol Chem. 254 (8): 2865–74. PMID 107164
↑ Huber R (1980). «Spatial structure of immunoglobulin molecules». Klin Wochenschr. 58 (22): 1217–31. PMID 6780722. doi:10.1007/BF01478928
↑ Heyman B (1996). «Complement and Fc-receptors in regulation of the antibody response». Immunol Lett. 54 (2-3): 195–9. PMID 9052877. doi:10.1016/S0165-2478(96)02672-7
↑ ab Ravetch J, Bolland S (2001). «IgG Fc receptors». Annu Rev Immunol. 19: 275–90. PMID 11244038. doi:10.1146/annurev.immunol.19.1.275
↑ Rus H, Cudrici C, Niculescu F (2005). «The role of the complement system in innate immunity». Immunol Res. 33 (2): 103–112. PMID 16234578. doi:10.1385/IR:33:2:103
↑ Racaniello, Vincent (6 de outubro de 2009). «Natural antibody protects against viral infection». Virology Blog. Consultado em 22 de janeiro de 2010. Cópia arquivada em 18 de novembro de 2010
↑ Milland J, Sandrin MS (Dezembro de 2006). «ABO blood group and related antigens, natural antibodies and transplantation». Tissue Antigens. 68 (6): 459–466. PMID 17176435. doi:10.1111/j.1399-0039.2006.00721.x
↑ Mian I, Bradwell A, Olson A (1991). «Structure, function and properties of antibody binding sites». J Mol Biol. 217 (1): 133–151. PMID 1988675. doi:10.1016/0022-2836(91)90617-F
↑ Fanning LJ, Connor AM, Wu GE (1996). «Development of the immunoglobulin repertoire». Clin. Immunol. Immunopathol. 79 (1): 1–14. PMID 8612345. doi:10.1006/clin.1996.0044
↑ ab Nemazee D (2006). «Receptor editing in lymphocyte development and central tolerance». Nat Rev Immunol. 6 (10): 728–740. PMID 16998507. doi:10.1038/nri1939
↑ Peter Parham. "The Immune System. 2nd ed. Garland Science: New York, 2005. pg.47–62
↑ Mraz, M.; Dolezalova, D.; Plevova, K.; Stano Kozubik, K.; Mayerova, V.; Cerna, K.; Musilova, K.; Tichy, B.; Pavlova, S.; Borsky, M.; Verner, J.; Doubek, M.; Brychtova, Y.; Trbusek, M.; Hampl, A.; Mayer, J.; Pospisilova, S. (2012). «MicroRNA-650 expression is influenced by immunoglobulin gene rearrangement and affects the biology of chronic lymphocytic leukemia». Blood. 119 (9): 2110–2113. PMID 22234685. doi:10.1182/blood-2011-11-394874
↑ ab Market, Eleonora; Papavasiliou, F. Nina (outubro de 2003). «V(D)J Recombination and the Evolution of the Adaptive Immune System». PLoS Biology. 1 (1): E16. PMC 212695. PMID 14551913. doi:10.1371/journal.pbio.0000016
↑ Bergman Y, Cedar H (2004). «A stepwise epigenetic process controls immunoglobulin allelic exclusion». Nat Rev Immunol. 4 (10): 753–761. PMID 15459667. doi:10.1038/nri1458
↑ Honjo T, Habu S (1985). «Origin of immune diversity: genetic variation and selection». Annu Rev Biochem. 54 (1): 803–830. PMID 3927822. doi:10.1146/annurev.bi.54.070185.004103
↑ ab Or-Guil M, Wittenbrink N, Weiser AA, Schuchhardt J (2007). «Recirculation of germinal center B cells: a multilevel selection strategy for antibody maturation». Immunol. Rev. 216: 130–41. PMID 17367339. doi:10.1111/j.1600-065X.2007.00507.x
↑ Neuberger M, Ehrenstein M, Rada C, Sale J, Batista F, Williams G, Milstein C (Março de 2000). «Memory in the B-cell compartment: antibody affinity maturation». Philos Trans R Soc Lond B Biol Sci. 355 (1395): 357–360. PMC 1692737. PMID 10794054. doi:10.1098/rstb.2000.0573
↑ Stavnezer J, Amemiya CT (2004). «Evolution of isotype switching». Semin. Immunol. 16 (4): 257–275. PMID 15522624. doi:10.1016/j.smim.2004.08.005
↑ Durandy A (2003). «Activation-induced cytidine deaminase: a dual role in class-switch recombination and somatic hypermutation». Eur. J. Immunol. 33 (8): 2069–2073. PMID 12884279. doi:10.1002/eji.200324133
↑ Casali P, Zan H (2004). «Class switching and Myc translocation: how does DNA break?». Nat. Immunol. 5 (11): 1101–1103. PMC 4625794. PMID 15496946. doi:10.1038/ni1104-1101
↑ Lieber MR, Yu K, Raghavan SC (2006). «Roles of nonhomologous DNA end joining, V(D)J recombination, and class switch recombination in chromosomal translocations». DNA Repair (Amst.). 5 (9–10): 1234–1245. PMID 16793349. doi:10.1016/j.dnarep.2006.05.013
↑ Weill, JC e outros: (1989). «Somatic hyperconversion diversifies the single VH gene of the chicken with a high incidence in the D region». Cell. 59
↑ Knight, KL: (1992). «Restricted VH gene usage and generation of antibody diversity in rabbit.». annu. Rev. immunol. 10
↑ page 22 in: Shoenfeld, Yehuda.; Meroni, Pier-Luigi.; Gershwin, M. Eric (2007). Autoantibodie. Amsterdam; Boston: Elsevier. ISBN 978-0-444-52763-9
↑ Spiess, Christoph; Zhai, Qianting; Carter, Paul J. (outubro de 2015). «Alternative molecular formats and therapeutic applications for bispecific antibodies». Molecular Immunology. 67 (2): 95–106. doi:10.1016/j.molimm.2015.01.003
↑ ab Farlex dictionary > polyvalent Citing: The American Heritage Medical Dictionary. 2004
↑ Gunasekaran K (2010). «Enhancing antibody Fc heterodimer formation through electrostatic steering effects: applications to bispecific molecules and monovalent IgG». The Journal of Biological Chemistry. 285 (25): 19637–19646. PMC 2885242. PMID 20400508. doi:10.1074/jbc.M110.117382
↑ Muller K.M (1998). «The first constant domain (CH1 and CL) of an antibody used as heterodimerization domain for bispecific miniantibodies». FEBS Letters. 422 (2): 259–264. doi:10.1016/s0014-5793(98)00021-0
↑ Gao C (1999). «Making artificial antibodies: A format for phage display of combinatorial heterodimeric arrays». PNAS. 96 (11): 6025–6030. PMC 26829. PMID 10339535. doi:10.1073/pnas.96.11.6025
↑ Litman, G; Cannon, JP y Dishaw, LJ: (novembro de 2005). «Reconstructing immune phylogeny:new perspectives». Nature. 5. doi:10.1038/nri1712 !CS1 manut: Nomes múltiplos: lista de autores (link)
↑ Bateman, A; Eddy, SR y Chothia, C (1996). «Members of the immunoglobulin superfamily in bacteria». Protein Science. 5 (5). PMID 8880921 !CS1 manut: Nomes múltiplos: lista de autores (link)
↑ Dersch P, Isberg RR. (2000). «An immunoglobulin superfamily-like domain unique to the Yersinia pseudotuberculosis invasin protein is required for stimulation of bacterial uptake via integrin receptors.». Infect Immun. 68 (5). PMID 10768991
↑ Matsunaga, J; Ko, AI e colaboradores: (2005). «Pathogenic Leptospira species express surface-exposed proteins belonging to the bacterial immunoglobulin superfamily». Mol Microbiol. 49 (4). PMCID PMC1237129 !CS1 manut: Nomes múltiplos: lista de autores (link)
↑ ateman A, Eddy SR, Mesyanzhinov VV: (1997). «A member of the immunoglobulin superfamily in bacteriophage T4». Virus Genes. 14 (2). PMID 9237357 !CS1 manut: Nomes múltiplos: lista de autores (link)
↑ Wojciechowicz D, Lu CF, Kurjan J, Lipke PN (1993). «Cell surface anchorage and ligand-binding domains of the Saccharomyces cerevisiae cell adhesion protein alpha-agglutinin, a member of the immunoglobulin superfamily». Mol Cell Biol. 13 (4). PMID 8455628 !CS1 manut: Nomes múltiplos: lista de autores (link)
↑ Grigorescu A, Chen MH, Zhao H, Kahn PC, Lipke PN (2000). «A CD2-based model of yeast alpha-agglutinin elucidates solution properties and binding characteristics». IUBMB Life. 50 (2). PMID 11185954 !CS1 manut: Nomes múltiplos: lista de autores (link)
↑ Nick Matzke (28 de abril de 2006). «Postulated intermediates in the molecular evolution of the Ig and TCR loci». Annotated Bibliography on the Evolutionary Origin of the Vertebrate Immune System. Journal of Experimental Medicine. Consultado em 22 de agosto de 2008
↑ Kubrycht J, Borecký J, Soucek P, Jezek P (2004). «Sequence similarities of protein kinase substrates and inhibitors with immunoglobulins and model immunoglobulin homologue: cell adhesion molecule from the living fossil sponge Geodia cydonium. Mapping of coherent database similarities and implications for evolution of CDR1 and hypermutation». Folia Microbiol (49). 3 PMID 15259763 !CS1 manut: Nomes múltiplos: lista de autores (link)
↑ Brümmendorf, T y Lemmon, V: (2001). «Immunoglobulin superfamily receptors: cis-interactions, intracellular adapters and alternative splicing regulate adhesion». Current opinion in cell biology. 13 (5). doi 10.1016/S0955-0674(00)00259-3 !CS1 manut: Nomes múltiplos: lista de autores (link)
↑ Strecker, G y otros: (2004). «Molecular recognition between glyconectins as an adhesion self-assembly pathway to multicellularity». J Biol Chem. 279 (15). PMID 14701844
↑ «The Evolution of Adaptative Immune Systems». Cell (124). 2006. DOI 10.1016/j.cell.2006.02.001
↑ Litman, GW e outros: (2006). «Genomic Insights into the Immune System of the Sea Urchin». Science. 314 (5801). DOI 10.1126/science.1134301
↑ Noverr, MC e Huffnagle, GB. «Does the microbiota regulate immune responses outside the gut?». Trends Microbiol. 12. PMID !CS1 manut: Nomes múltiplos: lista de autores (link)
↑ Oren M, Douek J, Fishelson Z, Rinkevich B: (2007). «Identification of immune-relevant genes in histoincompatible rejecting colonies of the tunicate Botryllus schlosseri». Dev Comp Immunol. 31 (9). PMID 17287019 !CS1 manut: Nomes múltiplos: lista de autores (link)
↑ Pancer Z, Diehl-Seifert B, Rinkevich B, Müller WE: (1997). «A novel tunicate (Botryllus schlosseri) putative C-type lectin features an immunoglobulin domain». DNA Cell Biol. 16 (6). PMID 9212174 !CS1 manut: Nomes múltiplos: lista de autores (link)
↑ Kapitonov, VV;Jurka, J: (2005). «RAG1 core and V(D)J recombination signal sequences were derived from Transib transposons». PLoS Biol. 3 !CS1 manut: Nomes múltiplos: lista de autores (link)
↑ Cannon JP, Haire RN, Litman GW: (2002). «Identification of diversified genes that contain immunoglobulin-like variable regions in a protochordate». Nat Immunol. 3 (12). PMID 12415263 !CS1 manut: Nomes múltiplos: lista de autores (link)
↑ Hernández Prada JA, Haire RN, Allaire M, Jakoncic J, Stojanoff V, Cannon JP, Litman GW, Ostrov DA: (2006). «Ancient evolutionary origin of diversified variable regions demonstrated by crystal structures of an immune-type receptor in amphioxus». Nature immunology. 7 (8). PMID !CS1 manut: Nomes múltiplos: lista de autores (link)
↑ Litman GW, Cannon JP, Dishaw LJ, Haire RN, Eason DD, Yoder JA, Prada JH, Ostrov DA: (2007). «Immunoglobulin variable regions in molecules exhibiting characteristics of innate and adaptive immune receptors». Immunol Res. 38 (1-3). PMID 17917037 !CS1 manut: Nomes múltiplos: lista de autores (link)
↑ Cooper, MD y Alder, MN (2006). «The Evolution of Adaptive Immune Systems». Cell. 124. DOI 10.1016/j.cell.2006.02.001 !CS1 manut: Nomes múltiplos: lista de autores (link)
↑ Kurt Buchmann (2014). «Evolution of Innate Immunity: Clues from Invertebrates via Fish to Mammals». Frontiers in Immunology. (5): 459. PMC 4172062. PMID 25295041. doi:10.3389/fimmu.2014.00459
↑ Janvier, P (1999). «Catching the first fish». Nature. 402
↑ ThomasBoehm (11 de setembro de 2012). «Evolution of Vertebrate Immunity». Current Biology. 22 (17): R722-R732
↑ Stein Tore Solem e Jørgen Stenvik. Antibody repertoire development in teleosts--a review with emphasis on salmonids and Gadus morhua L. Developmental & Comparative Immunology. Volume 30. Número 1-2, Antibody repertoire development, 2006. Páginas 57-76.
↑ J.D. Hansen, E.D. Landis and R.B. Phillips. Discovery of a unique Ig heavy-chain isotype (IgT) in rainbow trout: Implications for a distinctive B cell developmental pathway in teleost fish. Proceedings of the National Academy of Sciences U S A. Volume 102, Número 19, 2005. Páginas 6919-24.
↑ N. Danilova, J. Bussmann, K. Jekosch, L.A Steiner. The immunoglobulin heavy-chain locus in zebrafish: identification and expression of a previously unknown isotype, immunoglobulin Z. Nature Immunology, Volume 6. Número 3, 2005. Páginas 295-302.
↑ H. Dooley and M.F. Flajnik. Antibody repertoire development in cartilaginous fish. Developmental & Comparative Immunology. Volume 30. Número 1-2. Antibody repertoire development, 2006. Páginas 43-56.
↑ «Animated depictions of how antibodies are used in ELISA assays». Cellular Technology Ltd.—Europe. Consultado em 8 de maio de 2007. Cópia arquivada em 18 de novembro de 2010
↑ «Animated depictions of how antibodies are used in ELISPOT assays». Cellular Technology Ltd.—Europe. Consultado em 8 de maio de 2007. Cópia arquivada em 18 de novembro de 2010
↑ Stern P (2006). «Current possibilities of turbidimetry and nephelometry» (PDF). Klin Biochem Metab. 14 (3): 146–151. Arquivado do original (PDF) em 18 de novembro de 2010
↑ ab Dean, Laura (2005). «Chapter 4: Hemolytic disease of the newborn». Blood Groups and Red Cell Antigens. NCBI Bethesda (MD): National Library of Medicine (US),
↑ Rodriguez, Erik A.; Wang, Ye; Crisp, Jessica L.; Vera, David R.; Tsien, Roger Y.; Ting, Richard (27 de abril de 2016). «New Dioxaborolane Chemistry Enables [18F]-Positron-Emitting, Fluorescent [18F]-Multimodality Biomolecule Generation from the Solid Phase». Bioconjugate Chemistry (em inglês). 27 (5): 1390–1399. PMC 4916912. PMID 27064381. doi:10.1021/acs.bioconjchem.6b00164
↑ Feldmann M, Maini R (2001). «Anti-TNF alpha therapy of rheumatoid arthritis: what have we learned?». Annu Rev Immunol. 19: 163–96. PMID 11244034. doi:10.1146/annurev.immunol.19.1.163
↑ Doggrell S (2003). «Is natalizumab a breakthrough in the treatment of múltiple sclerosis?». Expert Opin Pharmacother. 4 (6): 999–1001. PMID 12783595. doi:10.1517/14656566.4.6.999
↑ Krueger G, Langley R, Leonardi C, Yeilding N, Guzzo C, Wang Y, Dooley L, Lebwohl M (2007). «A human interleukin-12/23 monoclonal antibody for the treatment of psoriasis». N Engl J Med. 356 (6): 580–92. PMID 17287478. doi:10.1056/NEJMoa062382 !CS1 manut: Nomes múltiplos: lista de autores (link)
↑ Plosker G, Figgitt D (2003). «Rituximab: a review of its use in non-Hodgkin's lymphoma and chronic lymphocytic leukaemia». Drugs. 63 (8): 803–43. PMID 12662126. doi:10.2165/00003495-200363080-00005
↑ Vogel C, Cobleigh M, Tripathy D, Gutheil J, Harris L, Fehrenbacher L, Slamon D, Murphy M, Novotny W, Burchmore M, Shak S, Stewart S (2001). «First-line Herceptin monotherapy in metastatic breast cancer». Oncology. 61 Suppl 2: 37–42. PMID 11694786. doi:10.1159/000055400 !CS1 manut: Nomes múltiplos: lista de autores (link)
↑ LeBien TW (2000). «Fates of human B-cell precursors». Blood. 96 (1): 9–23. PMID 10891425
↑ Ghaffer A (26 de março de 2006). «Immunization». Immunology - Chapter 14. University of South Carolina School of Medicine. Consultado em 6 de junho de 2007
↑ ab Fung Kee Fung K, Eason E, Crane J, Armson A, De La Ronde S, Farine D, Keenan-Lindsay L, Leduc L, Reid G, Aerde J, Wilson R, Davies G, Désilets V, Summers A, Wyatt P, Young D (2003). «Prevention of Rh alloimmunization». J Obstet Gynaecol Can. 25 (9): 765–73. PMID 12970812 !CS1 manut: Nomes múltiplos: lista de autores (link)
↑ Urbaniak S, Greiss M (2000). «RhD haemolytic disease of the fetus and the newborn». Blood Rev. 14 (1): 44–61. PMID 10805260. doi:10.1054/blre.1999.0123
↑ Tini M, Jewell UR, Camenisch G, Chilov D, Gassmann M (2002). «Generation and application of chicken egg-yolk antibodies». Comp. Biochem. Physiol., Part a Mol. Integr. Physiol. 131 (3): 569–574. PMID 11867282. doi:10.1016/S1095-6433(01)00508-6
↑ Cole SP, Campling BG, Atlaw T, Kozbor D, Roder JC (1984). «Human monoclonal antibodies». Mol. Cell. Biochem. 62 (2): 109–20. PMID 6087121. doi:10.1007/BF00223301
↑ Kabir S (2002). «Immunoglobulin purification by affinity chromatography using protein A mimetic ligands prepared by combinatorial chemical synthesis». Immunol Invest. 31 (3–4): 263–278. PMID 12472184. doi:10.1081/IMM-120016245
↑ ab Brehm-Stecher B, Johnson E (2004). «Single-cell microbiology: tools, technologies, and applications». Microbiol Mol Biol Rev. 68 (3): 538–559. PMC 515252. PMID 15353569. doi:10.1128/MMBR.68.3.538-559.2004. Cópia arquivada em 18 de novembro de 2010
↑ Williams N (2000). «Immunoprecipitation procedures». Methods Cell Biol. Methods in Cell Biology. 62: 449–453. ISBN 978-0-12-544164-3. PMID 10503210. doi:10.1016/S0091-679X(08)61549-6
↑ Kurien B, Scofield R (2006). «Western blotting». Methods. 38 (4): 283–293. PMID 16483794. doi:10.1016/j.ymeth.2005.11.007
↑ Scanziani E (1998). «Immunohistochemical staining of fixed tissues». Methods Mol Biol. 104: 133–140. ISBN 978-0-89603-525-6. PMID 9711649. doi:10.1385/0-89603-525-5:133
↑ Reen DJ. (1994). «Enzyme-linked immunosorbent assay (ELISA)». Methods Mol Biol. 32: 461–466. ISBN 0-89603-268-X. PMID 7951745. doi:10.1385/0-89603-268-X:461
↑ Kalyuzhny AE (2005). «Chemistry and biology of the ELISPOT assay». Methods Mol Biol. 302: 015–032. ISBN 1-59259-903-6. PMID 15937343. doi:10.1385/1-59259-903-6:015
↑ Saper (2005). «An open letter to our readers on the use of antibodies». Journal of Comparative Neurology. 493: 477–8. PMID 16304632. doi:10.1002/cne.20839
↑ «Implementing Rigor and Transparency in NIH & AHRQ Research Grant Applications»
↑ «On the reproducibility of science: unique identification of research resources in the biomedical literature». PeerJ. 2 de setembro de 2013. Consultado em 1 de setembro de 2014
↑ Bandrowski A, Brush M, Grethe JS, Haendel MA, Kennedy DN, Hill S, Hof PR, Martone ME, Pols M, Tan S, Washington N, Zudilova-Seinstra E, Vasilevsky N (2015). «The Resource Identification Initiative: a cultural shift in publishing». F1000Res. 4. 134 páginas. PMC 4648211. PMID 26594330. doi:10.12688/f1000research.6555.2
↑ «Reporting research antibody use: how to increase experimental reproducibility». F1000. 23 de agosto de 2013. Consultado em 1 de setembro de 2014
↑ «The Antibody Registry»
↑ «Resource Identification Initiative». FORCE11. Consultado em 18 de abril de 2016
↑ Tini M, Jewell UR, Camenisch G, Chilov D, Gassmann M (2002). «Generation and application of chicken egg-yolk antibodies». Comp. Biochem. Physiol., Part a Mol. Integr. Physiol. 131 (3): 569–74. PMID 11867282 !CS1 manut: Nomes múltiplos: lista de autores (link)
↑ Cole SP, Campling BG, Atlaw T, Kozbor D, Roder JC (1984). «Human monoclonal antibodies». Mol. Cell. Biochem. 62 (2): 109–20. PMID 6087121 !CS1 manut: Nomes múltiplos: lista de autores (link)
↑ Kabir S (2002). «Immunoglobulin purification by affinity chromatography using protein A mimetic ligands prepared by combinatorial chemical synthesis». Immunol Invest. 31 (3-4): 263–78. PMID 12472184. doi:10.1081/IMM-120016245
↑ Lennard, S (2001). «Standard Protocols for the Construction of scFv Libraries». Springer protocols. DOI 10.1385/1-59259-240-6:059
↑ Altria, KD (1996). Capillary Electrophoresis Guidebook Principles, Operation, and Applications. [S.l.]: Humana Press. p. 226. ISBN 1-59259-538-3
↑ Blackburn, GM y colaboradores: (1996). «Toward antibody-directed "abzyme" prodrug therapy, ADAPT: carbamate prodrug activation by a catalytic antibody and its in vitro application to human tumor cell killing» (PDF). PNAS. 93 (2)
↑ Shiro Kobayashi, Helmut Ritter, David Kaplan (2006). Enzyme-Catalyzed Synthesis of Polymers,. [S.l.]: Birkhäuser. 206 páginas. ISBN 3-540-29212-8 !CS1 manut: Nomes múltiplos: lista de autores (link)
↑ Hamers-Casterman C, Atarhouch T, Muyldermans S, Robinson G, Hamers C, Songa EB, Bendahman N, Hamers R., Naturally occurring antibodies devoid of light chains, Nature. 1993 junho 3;363(6428):446-8
↑ «Nanobodies herald a new era in cancer therapy». Medical News. 2004
↑ Cano, AA. «ANTICUERPOS TERAPEUTICOS: EL CASO DE LOS ANTIVENENOS» (PDF). Sociedade Mexicana de Bioquímica. Consultado em 25 de agosto de 2008. Cópia arquivada (PDF) em 3 de dezembro de 2008
↑ Arquivado em 18 de novembro de 2010 no WebCite
WAM
↑ Marcatili P, Rosi A, Tramontano A (2008). «PIGS: automatic prediction of antibody structures». Bioinformatics. 24 (17): 1953–1954. PMID 18641403. doi:10.1093/bioinformatics/btn341. Cópia arquivada em 18 de novembro de 2010
Prediction of Immunoglobulin Structure (PIGS)
↑ Arquivado em 18 de novembro de 2010 no WebCite
RosettaAntibody
↑ Park, Hyeongsu. «Written Description Problems of the Monoclonal Antibody Patents after Centocor v. Abbott». jolt.law.harvard.edu. Consultado em 12 de dezembro de 2014
↑ Pham, V. et al. De novo proteomic sequencing of a monoclonal antibody raised against OX40 ligand. Anal. Biochem. 352, 77–86 (2006).
↑ Ma, B. et al. PEAKS: Powerful Software for Peptide De Novo Sequencing by Tandem Mass Spectrometry. Rapid Commun.Mass Spectrom. 17(20), 2337–2342 (2003).
↑ Zhang, J. et al. PEAKS DB: De Novo Sequencing Assisted Database Search for Sensitive and Accurate Peptide Identification. Mol. Cell. Proteomics 10.1074/mcp.M111.010587 (2011).
↑ Cottrell, J. S. & London, U. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20(18), 3551–3567 (1999).
↑ Bandeira, N., Tang, H., Bafna, V. & Pevzner, P. Shotgun protein sequencing by tandem mass spectra assembly. Anal. Chem. 76, 7221–7233 (2004).
↑ Liu, X., Han, Y., Yuen, D. & Ma, B. Automated protein (re)sequencing with MS/MS and a homologous database yields almost full coverage and accuracy. Bioinformatics. 25(17), 2174–2180 (2009).
↑ Liu, X. et al. De novo protein sequencing by combining top-down and bottom-up tandem mass spectra. J. Proteome Res. 13(7), 3241–3248 (2014).
↑ Tran, N.H. et al. Complete De Novo Assembly of Monoclonal Antibody Sequences. Scientific Reports. 6(31730). (2016).
Controle de autoridade |
|
|---|